
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,792
- Solutions
- 3
- Reaction score
- 3,053
- Points
- 113
- Deals
- 1
Введение
Лизергиновая кислота, основной фрагмент, получаемый из алкалоидов спорыньи, была синтезирована в четырнадцатой последовательности, начиная с 3-бета-карбоксиэтилиндола. Исходный материал был преобразован в промежуточный 1-бензоил-5-кетон-1,2,2a-3,4,5-гексагидробенз-[cd]-индол (3), содержащий три из четырех колец, присутствующих в лизергиновой кислоте. Этот кетон, в свою очередь, превращается в тетрациклическое соединение, 9-кето-7-метил-4,5,5a,6,6a,7,8,9-октагидроиндоло-[4.3-fe]-хинолин (8), а затем в лизергиновую кислоту (14). Этот синтез не прост и требует большого лабораторного опыта и знаний химии. Кроме того, здесь есть несколько манипуляций с опасными веществами, которые должны проводиться с соблюдением строгих мер безопасности.
Температура кипения: 536,2±50,0 °C при 760 мм рт. ст.;
Температура плавления: 240 °C;
Молекулярная масса: 268,31 г/моль;
Плотность: 1,4 ± 0,1 г/мл;
CAS Number: 82-58-6.
Оборудование и стеклянная посуда:
- Стальной реактор для гидрирования 2-3 л;
- Стальной автоклав 500 мл;
- Лабораторные весы (0,01 - 500 г);
- Круглодонные колбы 100, 200, 500 мл, 5 и 10 л;
- Компрессор для получения водорода (H2) и его происхождение;
- Колба Бюхнера и воронка (большая) 5 л [для небольших количеств можно использовать фильтр Шотта];
- Ротовап машина (большая);
- Источник вакуума;
- Воронки для разделения 500 мл и 2 л;
- Баллон с азотом ~50-70 л (1 бар);
- Пробки с перегородками для колб;
- Баня с соленой ледяной водой;
- 5 л x2, 2 л x2; 1 л x2; 500 мл x2; 100 мл x3; 50 мл x2 Мензурки;
- Стеклянный шприц или пипетка Пастера;
- Магнитная мешалка или верхняя мешалка;
- Установка для вакуумной дистилляции;
- Рефлюксный конденсатор;
- Подставка для реторты и зажим для крепления аппарата;
- Лабораторный термометр (от -20 °C до 200 °C) с адаптером для колбы;
- Индикаторная бумага для определения pH;
- Стеклянная палочка и шпатель;
- лампочка мощностью 250 Вт.
Реактивы.
- 3-Индолпропионовая кислота (1), 94,6 г (0,5 моль);
- 9,5 л дистиллированной воды (H2O);
- ~400 г гидроксида натрия (NaOH);
- 116 г никеля Рани (Ni);
- 1050 мл Соляная (HCl) кислота концентрированная;
- 2 мл серной кислоты (H2SO4 конц.);
- 210 мл 12N раствора гидроксида натрия (NaOH);
- 180 мл Бензоил хлорида;
- ~1,5 л метанола (MeOH);
- ~1,6 л этанола (EtOH);
- 201,2 мл Тионилхлорид (SOCl2);
- 1950 мл Дисульфид углерода (CS2);
- 240 г хлорида алюминия (AlCl3);
- 2,5 л бензола;
- 500 мл 2N гидроксида натрия (NaOH);
- ~3,2 л Диэтиловый эфир (Et2O);
- 3,3 л ледяной уксусной кислоты (AcOH);
- 352 г (1,1 моль) пиридингидробромида пербромида;
- 5 л Хлороформ (CHCl3);
- ~1000 г Сульфат магния (MgSO4);
- 307 г (2,35 моль) Метиламиноацетон этиленкеталь (5);
- 4,5 л Бензол;
- ~500 г Активированный уголь (C);
- ~1 л Ацетон;
- ~500 г Бикарбонат натрия (NaHCO3);
- 80 мл Холодный уксусный ангидрид (Ac2O);
- 1,5 г борогидрида натрия (NaBH4);
- 75 мл Диоксид серы (SO2 жидкий);
- 40 г цианида натрия (порошок NaCN);
- 300 мл цианистого водорода (HCN жидкость);
- 78 мл 1,5% раствора гидроксида калия (KOH);
- 8,5 г гидратированного арсената натрия;
- ~ 50 мл Ксилол;
- 100 мл разбавленного раствора гидроксида аммония (NH4OH);
- 16,9 г метоксида натрия (MeONa).
Процедура
1-Бензоил-3-(бета-карбоксиэтил)-2,3-дигидроиндол (2)3-Индолпропионовую кислоту (1), 94,6 г (0,5 моль), растворили в 600 мл воды, содержащей 20 г гидроксида натрия. Раствор смешивали с примерно 100 г никелевого катализатора Raney и гидрировали при RT в стальной гидрирующей бомбе объемом 2-3 л при давлении 3000-4000 фунтов на квадратный дюйм (207-276 бар) H2. Восстановление обычно завершается за 20-30 ч, после чего катализатор отфильтровывают и промывают небольшим количеством воды. К фильтрату добавляли 85 мл концентрированной кислоты HCl и раствор охлаждали. Если восстановление было неполным, то на этом этапе отделялась непрореагировавшая индолпропионовая кислота, которую удаляли фильтрованием. Затем фильтрат бензоилировали по обычной процедуре Шоттена-Баумана, используя 210 мл 12N гидроксида натрия и 180 мл бензоилхлорида. Раствор поддерживали щелочным на протяжении всего процесса бензоилирования, а температуру поддерживали ниже 40 °C путем охлаждения. Когда бензоилхлорид полностью прореагировал, смесь охладили и подкислили 300 мл концентрированной HCl. Неочищенный продукт отфильтровали и промыли водой, после чего экстрагировали 4 х 1 л горячей воды для удаления бензойной кислоты. Горячий сиропообразный продукт (2), после декантации водного экстракта, был кристаллизован из нескольких объемов метанола; выход 103 г (70 %), MP: 151-153 °C.
1-Бензоил-5-кето-1,2,2а,3,4,5-гексагидробенз-[cd]-индол (3)
1-Бензоил-3-(бета-карбоксиэтил)-2,3-дигидроиндол (2), 118 г (0,4 моль), смешивали с 200 мл чистого тионилхлорида. Раствору дали отстояться в течение 30 мин, после чего осторожно нагревали на паровой бане в течение 15-20 мин. Избыток тионилхлорида полностью выпаривали при температуре ниже 30 °C в вакууме, а неочищенный хлорид кислоты растворяли в 200 мл дисульфида углерода. Раствор хлорида кислоты был добавлен тонкой струйкой к энергично перемешиваемой суспензии 240 г хлорида алюминия в 1750 мл дисульфида углерода, находящейся в 5-литровой колбе (в капотнице!!!). Отделился комплекс, и перемешивание стало затруднительным. Смесь нагревали под флюсом и перемешивали в течение часа для завершения реакции, после чего очень осторожно разложили, добавив 500 г льда, 250 мл конц. кислоты HCl и 500 мл воды. Во время разложения поддерживалось перемешивание, охлаждение происходило путем периодической отгонки дисульфида углерода in vacuo, и продукт экстрагировали 2 л бензола. Экстракт тщательно промыли тремя порциями 500 мл 2N гидроксида натрия, а затем водой. Его сушили над сульфатом магния и выпаривали до небольшого объема в вакууме. Медленное добавление нескольких объемов эфира привело к кристаллизации желтого кетона (3) . Его отфильтровали и промыли эфиром; выход 85,3 г (77 %), тпл: 146-147 °C. Образец был перекристаллизован для анализа из бензол-эфира.
1-Бензоил-3-(бета-карбоксиэтил)-2,3-дигидроиндол (2), 118 г (0,4 моль), смешивали с 200 мл чистого тионилхлорида. Раствору дали отстояться в течение 30 мин, после чего осторожно нагревали на паровой бане в течение 15-20 мин. Избыток тионилхлорида полностью выпаривали при температуре ниже 30 °C в вакууме, а неочищенный хлорид кислоты растворяли в 200 мл дисульфида углерода. Раствор хлорида кислоты был добавлен тонкой струйкой к энергично перемешиваемой суспензии 240 г хлорида алюминия в 1750 мл дисульфида углерода, находящейся в 5-литровой колбе (в капотнице!!!). Отделился комплекс, и перемешивание стало затруднительным. Смесь нагревали под флюсом и перемешивали в течение часа для завершения реакции, после чего очень осторожно разложили, добавив 500 г льда, 250 мл конц. кислоты HCl и 500 мл воды. Во время разложения поддерживалось перемешивание, охлаждение происходило путем периодической отгонки дисульфида углерода in vacuo, и продукт экстрагировали 2 л бензола. Экстракт тщательно промыли тремя порциями 500 мл 2N гидроксида натрия, а затем водой. Его сушили над сульфатом магния и выпаривали до небольшого объема в вакууме. Медленное добавление нескольких объемов эфира привело к кристаллизации желтого кетона (3) . Его отфильтровали и промыли эфиром; выход 85,3 г (77 %), тпл: 146-147 °C. Образец был перекристаллизован для анализа из бензол-эфира.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
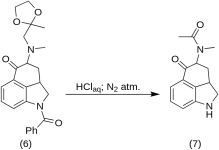
1-Бензоил-2,2а,3,4-тетрагидро-4-[метил-(2-метил-1,2-диоксолан-2-ил-метил)-амино]-бенз-[cd]-индол-5-(1H)-он (6)
Раствор 270 г (0,76 моль) 1-бензоил-4-бром-5-кето-1,2,2а,3,4,5-гексагидробенз-[cd]-индола(4) и 307 г (2.35 моль) метиламиноацетон-этиленового кеталя (5) в 4500 мл сухого бензола в 10 л RBF с рефлюкс-конденсатором под азотом в течение 21 ч. Смесь охладили, и отфильтровали 151 г (93,5 %) гидробромида метиламиноацетона этиленкеталя, MP: 158-159 °C.
Фильтрат промыли несколько раз водой со льдом, после чего экстрагировали 2,5 л холодной разбавленной HCl, содержащей 150 мл концентрированной кислоты. Кислотные экстракты были немедленно добавлены к избытку ледяного разбавленного гидроксида натрия. Продукт экстрагировали 1 л хлороформа, хлороформный раствор высушили над сульфатом магния, обработали углеродом и сконцентрировали в вакууме. Остаточный кеталь-кетон (6) кристаллизовали из ацетона; МР: и МР смеси: 135-136 °C, выход 220 г (71 %).
5-Кето-4-[N-метил-N-ацетониламино]-1,2,2а,3,4,5-гексагидробенз-[cd]-индол (7)
20 г 1-бензоил-2,2а,3,4-тетрагидро-4-[метил-(2-метил-1, 6) растворили в смеси 250 мл концентрированной кислоты HCl и 250 мл воды, и выдерживали под азотом при 37 °C в 3-5 л RBF в течение пяти дней. Смесь охладили, обработали углеродом, отфильтровали и фильтрат сконцентрировали in vacuo до небольшого объема. Остаток обрабатывали избытком бикарбоната натрия, продукт экстрагировали холодным хлороформом и удаляли растворитель в вакууме при RT. Неочищенный дикетон (7) измельчили в порошок, смешали с 75 мл бензол-эфира 1:1 и отфильтровали; выход 9,8 г (77 %), MP: 105-107 °C. Образец для анализа был перекристаллизован из бензол-эфира или этанола; МР: 109-110 °C; моногидрохлорид был получен из разбавленного этанола; МР: 200 °C дек.
9-Кето-7-метил-4,5,5а,6,6а,7,8,9-октагидроиндоло-[4,3-fg]-хинолин (8)
25 г 5-Кето-4-[N-метил-N-ацетонил]-амино-1,2,2а,3,4,5-гексагидробенз-[cd]-индола (7) смешивали с 550 мл абсолютного этанола. Смесь перемешивали под азотом и охлаждали до -15 °C в 2-5 л RBF. Затем добавили метоксид натрия, 16,9 г, и смесь перемешивали при температуре от -10 °C до -12 °C в течение десяти минут. Реакционную смесь охладили до -25 °C, продукт отфильтровали на 6,5-дюймовой воронке Бюхнера и промыли небольшим количеством холодного этанола и эфира. При минимальном воздействии воздуха (содержит метоксид натрия!) неочищенный кетон (8) был немедленно просушен с небольшим количеством ледяной воды и повторно отфильтрован. Его промывали ледяной водой, этанолом и эфиром; выход 16,2 г (69 %), MP: 145-147 °C. Аналитический образец был перекристаллизован из разбавленного этанола; MP: 155-157 °C; Дигидрохлорид был получен и перекристаллизован из водного ацетона; MP: 270 °C dec.
4-Ацетил-9-кето-7-метил-4,5,5a,6,6a,7,8,9-октагидроиндоло-[4,3-fg]-хинолин (9)
9-кето-7-метил-4,5,5a,6,6a,7,8,9-октагидроиндоло-[4,3-fg]-хинолин (8), 24 г, был добавлен к 80 мл холодного уксусного ангидрида. Смесь выдерживали при 25 °C в 200 мл RBF около 5 мин, после чего тщательно охладили, и продукт (9) отфильтровали и промыли эфиром; выход 20,5 г (76 %), mp: 167-170 °C. Вторая культура была получена выпариванием фильтрата, что увеличило общий выход до 82 %. Образец был перекристаллизован из ацетон-этанола; MP: 169-170 °C; Гидрохлорид был получен в этаноле и перекристаллизован из водного этанола; MP: 250 °C дек.
9-кето-7-метил-4,5,5a,6,6a,7,8,9-октагидроиндоло-[4,3-fg]-хинолин (8), 24 г, был добавлен к 80 мл холодного уксусного ангидрида. Смесь выдерживали при 25 °C в 200 мл RBF около 5 мин, после чего тщательно охладили, и продукт (9) отфильтровали и промыли эфиром; выход 20,5 г (76 %), mp: 167-170 °C. Вторая культура была получена выпариванием фильтрата, что увеличило общий выход до 82 %. Образец был перекристаллизован из ацетон-этанола; MP: 169-170 °C; Гидрохлорид был получен в этаноле и перекристаллизован из водного этанола; MP: 250 °C дек.
4-Ацетил-9-гидрокси-7-метил-4,5,5а,6,6а,7,8,9-октагидроиндоло-[4,3-fg]-хинолин (10)
10 г 4-ацетил-9-кето-7-метил-4,5,5а,6,6a,7,8,9-октагидроксиндоло-[4,3-fg]-хинолина (9) добавили к смеси 150 мл метанола и 10 мл воды в 500 мл RBF. Добавили 1,5 г борогидрида натрия, дали реакции протекать при RT до небольшого объема и добавили смесь 15 мл конц. кислоты HCl и 60 мл воды. Выделившийся при охлаждении гидрохлорид (10) отфильтровали и промыли метанолом, 9,0 г (79 %). Образец был перекристаллизован из разбавленного этанола; MP: 245-246 °C дек.
4-Ацетил-9-хлор-7-метил-4,5,5а,6,6а,7,8,9-октагидроиндоло-[4,3-fg]-хинолина гидрохлорид (11)
4-Ацетил-9-гидрокси-7-метил-4,5,5а,6,6а,7,8,9-октагидроиндоло-[4,3-fg]-хинолина гидрохлорид (10), 3.1 г растворяли в 75 мл жидкого диоксида серы, помещенного в стеклянный вкладыш в стальном автоклаве объемом 500 мл. Добавили 1,2 мл тионилхлорида, герметично закрыли сосуд и выдерживали при 25 °C в течение 6 ч. Автоклав проветрили и удалили реакционную смесь. Диоксиду серы дали испариться, а объем раствора поддерживали постоянным путем медленного добавления сухого эфира. Аморфный гидрохлорид хлора (11) отфильтровали, промыли эфиром и высушили в вакууме, MP: 130-135 °C dec. Выход 3,5 г. Использование 9-бета-эпимерного спирта в этой реакции дало тот же хлорид с сопоставимым выходом.
4-Ацетил-9-циано-7-метил-4,5,5a,6,6a,7,8,9-октагидроиндоло-[4,3-fg]-хинолин (12)
Сухой порошкообразный цианид натрия, 40 г, был добавлен к 300 мл ледяного жидкого цианистого водорода. Смесь перемешивали, охлаждали во льду и добавили 7,5 г неочищенного аморфного 4-ацетил-9-хлор-7-метил-4,5,5а,6,6а,7,8,9-октагидроиндоло [4,3f/g]-хинолина гидрохлорида (11) , описанного выше. Перемешивание продолжалось в 500 мл RBF в течение 30 мин, после чего цианистый водород был быстро отогнан под пониженным давлением при температуре около 10 °C. Остаток смешивали с хлороформом и ледяной водой, и полученную смесь фильтровали. Органический слой отделили, а водную фазу дважды экстрагировали хлороформом. Объединенные экстракты сушили над сульфатом магния, обесцвечивали и отгоняли растворитель in vacuo. Продукт (12) был кристаллизован из этилацетата; выход 3,3 г. (54% от общего количества на основе гидрохлорида спирта), м.п. 172-174 °C. Перекристаллизация из того же растворителя повысила м.п. до 181-182 °C.
Сухой порошкообразный цианид натрия, 40 г, был добавлен к 300 мл ледяного жидкого цианистого водорода. Смесь перемешивали, охлаждали во льду и добавили 7,5 г неочищенного аморфного 4-ацетил-9-хлор-7-метил-4,5,5а,6,6а,7,8,9-октагидроиндоло [4,3f/g]-хинолина гидрохлорида (11) , описанного выше. Перемешивание продолжалось в 500 мл RBF в течение 30 мин, после чего цианистый водород был быстро отогнан под пониженным давлением при температуре около 10 °C. Остаток смешивали с хлороформом и ледяной водой, и полученную смесь фильтровали. Органический слой отделили, а водную фазу дважды экстрагировали хлороформом. Объединенные экстракты сушили над сульфатом магния, обесцвечивали и отгоняли растворитель in vacuo. Продукт (12) был кристаллизован из этилацетата; выход 3,3 г. (54% от общего количества на основе гидрохлорида спирта), м.п. 172-174 °C. Перекристаллизация из того же растворителя повысила м.п. до 181-182 °C.
9-Карбометокси-7-метил-4,5,5a,6,6a,7,8,9-октагидроиндоло-[4,3-fg]-хинолин (13)
Продукт (12) , указанный выше, 1,0 г, смешивали с 15 мл метанола и 0,25 мл воды. Смесь охладили и медленно добавили 2 мл концентрированной серной кислоты. Раствор герметично закрыли в стеклянной трубке под азотом и нагревали при 100 °C в течение 23-24 ч в 100 мл RBF с рефлюкс-конденсатором. Смесь обрабатывали обесцвеченным углем и затем концентрировали in vacuo до объема около 10 мл. Ее выливали на смесь хлороформа (30 мл), льда и 10 г бикарбоната натрия. Хлороформный слой отделили, а водную фазу экстрагировали 3 х 10 мл хлороформа. Объединенные экстракты сушили над сульфатом магния, выпаривали досуха, и продукт (13) кристаллизовали из бензола; выход 0,51 г (53 %), тпл: 159-160 °C. Он был перекристаллизован из этилацетата; MP: 160-161 °C.
Продукт (12) , указанный выше, 1,0 г, смешивали с 15 мл метанола и 0,25 мл воды. Смесь охладили и медленно добавили 2 мл концентрированной серной кислоты. Раствор герметично закрыли в стеклянной трубке под азотом и нагревали при 100 °C в течение 23-24 ч в 100 мл RBF с рефлюкс-конденсатором. Смесь обрабатывали обесцвеченным углем и затем концентрировали in vacuo до объема около 10 мл. Ее выливали на смесь хлороформа (30 мл), льда и 10 г бикарбоната натрия. Хлороформный слой отделили, а водную фазу экстрагировали 3 х 10 мл хлороформа. Объединенные экстракты сушили над сульфатом магния, выпаривали досуха, и продукт (13) кристаллизовали из бензола; выход 0,51 г (53 %), тпл: 159-160 °C. Он был перекристаллизован из этилацетата; MP: 160-161 °C.
Синтетическая dl-лизергиновая кислота (14)
Смесь 9-карбометокси-7-метил-4,5,5a,6,6a,7,8,9-октагидроиндоло-[4,3-fg]-хинолина (13), 3,9 г, и 78 мл 1,5% раствора гидроксида калия подвергали рефлюксу в течение 30 мин под азотом. Добавили гидратированный арсенат натрия, 8,5 г, и никель Рэни (16 г, влажный), предварительно дезактивированный кипячением в суспензии ксилола, и смесь нагревали под рефлюксом и перемешивали в атмосфере азота в течение 20 часов в 200 мл RBF с рефлюкс-конденсатором. Раствор обрабатывали углеродом, и сырая лизергиновая кислота (14) выпадала в осадок при нейтрализации до pH 5,6. Ее отфильтровали и промыли водой; выход 1,04 г, MP: 240-242 °C dec. Также была получена вторая культура, 0,16 г, MP: 233-235 °C dec.; общий выход 30%. Кислоту можно было очистить, растворив ее в разбавленном гидроксиде аммония, обработав обесцвечивающим углем и переосадив диоксидом углерода, MP: 242-243 °C dec; смесь с dl-лизергиновой кислотой, полученной из природной d-лизергиновой кислоты, также была 242-243 °C dec. Безводная кислота была получена сушкой in vacuo в течение нескольких часов при 150 °C.
Смесь 9-карбометокси-7-метил-4,5,5a,6,6a,7,8,9-октагидроиндоло-[4,3-fg]-хинолина (13), 3,9 г, и 78 мл 1,5% раствора гидроксида калия подвергали рефлюксу в течение 30 мин под азотом. Добавили гидратированный арсенат натрия, 8,5 г, и никель Рэни (16 г, влажный), предварительно дезактивированный кипячением в суспензии ксилола, и смесь нагревали под рефлюксом и перемешивали в атмосфере азота в течение 20 часов в 200 мл RBF с рефлюкс-конденсатором. Раствор обрабатывали углеродом, и сырая лизергиновая кислота (14) выпадала в осадок при нейтрализации до pH 5,6. Ее отфильтровали и промыли водой; выход 1,04 г, MP: 240-242 °C dec. Также была получена вторая культура, 0,16 г, MP: 233-235 °C dec.; общий выход 30%. Кислоту можно было очистить, растворив ее в разбавленном гидроксиде аммония, обработав обесцвечивающим углем и переосадив диоксидом углерода, MP: 242-243 °C dec; смесь с dl-лизергиновой кислотой, полученной из природной d-лизергиновой кислоты, также была 242-243 °C dec. Безводная кислота была получена сушкой in vacuo в течение нескольких часов при 150 °C.
Attachments

Last edited:
