2C-B
4-BROMO-2,5-DIMETOXIFENETILAMINA
SÍNTESE: Uma solução de 100 g de 2,5-dimetoxibenzaldeído em 220 g de nitrometano foi tratada com 10 g de acetato de amónio anidro e aquecida num banho de vapor durante 2,5 h, com agitação ocasional. A mistura reacional, de cor vermelha intensa, foi removida do excesso de nitrometano sob vácuo e o resíduo cristalizou espontaneamente. Este nitroestireno bruto foi purificado por trituração sob IPA, filtragem e secagem ao ar, para obter 85 g de 2,5-dimetoxi-beta-nitroestireno como um produto amarelo-alaranjado de pureza adequada para a etapa seguinte. Pode obter-se uma purificação adicional por recristalização a partir de IPA em ebulição.
Num balão de fundo redondo de 2 L equipado com um agitador magnético e colocado sob uma atmosfera inerte, foram adicionados 750 mL de THF anidro, contendo 30 g de LAH. Em seguida, adicionou-se, em solução de THF, 60 g de 2,5-dimetoxi-beta-nitrostyrene. A solução final apresentava uma cor amarelo-castanho sujo e foi mantida à temperatura de refluxo durante 24 h. Após arrefecimento, o excesso de hidreto foi destruído pela adição gota a gota de IPA. Em seguida, foram adicionados 30 mL de NaOH a 15% para converter os sólidos inorgânicos numa massa filtrável. A mistura reacional foi filtrada e o bolo filtrante foi lavado primeiro com THF e depois com MeOH. Os licores-mãe e as lavagens combinados foram libertados do solvente sob vácuo e o resíduo foi suspenso em 1,5 L de H2O. Este foi acidificado com HCl, lavado com 3x100 mL de CH2Cl2, tornado fortemente básico com NaOH a 25% e reextraído com 4x100 mL de CH2Cl2. Os extractos combinados foram removidos do solvente sob vácuo, obtendo-se 26 g de resíduo oleoso, que foi destilado a 120-130 °C a 0,5 mm/Hg para dar 21 g de um óleo branco, 2,5-dimetoxifenetilamina
(2C-H), que capta muito rapidamente o dióxido de carbono do ar.
A uma solução bem agitada de 24,8 g de 2,5-dimetoxifenetilamina em 40 mL de ácido acético glacial, foram adicionados 22 g de bromo elementar dissolvido em 40 mL de ácido acético. Após alguns minutos, verificou-se a formação de sólidos e a evolução simultânea de calor considerável. A mistura reacional foi deixada à temperatura ambiente, filtrada e os sólidos foram lavados com moderação com ácido acético frio. Este era o sal de bromidrato. Existem muitas formas complicadas de sal, tanto polimorfos como hidratos, que podem tornar o isolamento e a caraterização do 2C-B traiçoeiros. O caminho mais feliz é formar o sal cloridrato insolúvel por meio da base livre. Toda a massa de sal molhado em ácido acético foi dissolvida em H2O quente, tornada básica a pelo menos pH 11 com NaOH a 25% e extraída com 3x100 mL de CH2Cl2. A remoção do solvente deu 33,7 g de resíduo que foi destilado a 115-130 °C a 0,4 mm/Hg. O óleo branco, 27,6 g, foi dissolvido em 50 mL de H2O contendo 7,0 g de ácido acético. Esta solução clara foi agitada vigorosamente e tratada com 20 mL de HCl concentrado. Verificou-se a formação imediata do sal anidro do cloridrato de 2,5-dimetoxi-4-bromofenetilamina (2C-B). Esta massa de cristais foi removida por filtração (pode ser solta consideravelmente pela adição de mais 60 mL de H2O), lavada com um pouco de H2O, e depois com várias porções de 50 mL de Et2O. Quando completamente seco ao ar, foram obtidos 31,05 g de agulhas brancas finas, com um mp de 237-239 °C com decomposição. Quando há demasiado H2O presente no momento da adição do HCl concentrado final, obtém-se uma forma hidratada de 2C-B. O sal de bromidrato funde a 214,5-215 °C. O sal de acetato tem uma temperatura de fusão de 208-209 °C.
DOSAGEM: 12 - 24 mg.
DURAÇÃO: 4 - 8 h.
DOM
STP; 2,5-DIMETOXI-4-METILANFETAMINA
SÍNTESE: A uma solução de 54,9 g de 2,5-dimetoxi-4-metilbenzaldeído (ver a receita de
2C-D para a sua preparação) em 215 g de ácido acético glacial, adicionaram-se 19,5 g de acetato de amónio anidro e 30,6 g de nitroetano. Esta mistura foi aquecida durante 3 h no banho de vapor, a mistura reacional foi arrefecida num banho de gelo húmido, permitindo a formação espontânea de cristais amarelos. Foi adicionada a maior quantidade possível de H2O (apenas para evitar um carácter oleoso turvo persistente) e, após algumas horas adicionais de repouso, o 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropeno cristalino foi removido por filtração e recristalizado a partir de ácido acético em ebulição. O rendimento, após secagem até peso constante, foi de 28,3 g e a temperatura média de 87-88 °C. Anal. (C12H15NO4) C, H ,N.
Uma suspensão de 9,5 g de LAH em 750 mL de Et2O anidro bem agitado foi mantida em refluxo sob uma atmosfera inerte, com o retorno do solvente condensado passando por um dedal de Soxhlet contendo 9,5 g de 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropeno. Após a adição completa do nitroestireno, a suspensão agitada foi mantida em refluxo durante mais 4 horas, depois arrefecida à temperatura ambiente e deixada a agitar durante a noite. O excesso de hidreto foi destruído pela adição de 750 mL de H2SO4 a 8%, cautelosamente, até que a evolução do hidrogénio cessasse e, em seguida, a uma velocidade que permitisse a dispersão dos sólidos formados. As fases foram separadas, a fase aquosa foi lavada uma vez com Et2O, tratada com 225 g de tartarato de sódio e potássio e, finalmente, tornada básica (pH >9) com NaOH a 5%. Esta fase foi extraída com 3x150 mL de CH2Cl2, os extractos foram reunidos e o solvente foi removido sob vácuo. O resíduo foi 9,6 g de um óleo límpido que formou espontaneamente cristais com uma temperatura de 60,5-61 °C a partir de hexano. Estes sólidos foram dissolvidos em 150 mL de Et2O anidro e saturados com gás HCl anidro. Após repouso à temperatura ambiente durante 2 h, o cloridrato cristalino de 2,5-dimetoxi-4-metilanfetamina (DOM) foi removido por filtração, lavado com Et2O e seco ao ar até peso constante. Obteve-se 8,25 g de cristais brancos brilhantes que tinham um PM de 190,5-191,5 °C. O sulfato tinha uma temperatura de 131 °C. Anal. (C12H20ClNO2) C, H ,N.
O nitroestireno acima referido pode também ser convertido no produto final amina através do intermediário da fenilacetona correspondente. A uma suspensão bem agitada de 10,4 g de ferro em pó em 20 mL de ácido acético glacial mantida à temperatura de refluxo, foram adicionados 4,9 g de 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropeno como um sólido. O refluxo foi continuado durante 2 h e depois tudo foi filtrado através de Celite húmida. Após lavagem com 300 mL de H2O seguida de 300 mL de Et2O, o filtrado combinado e as lavagens foram separados e a fase aquosa extraída com 2x100 mL de Et2O. A fase orgânica e os extractos foram combinados e lavados com 2x100 mL de K2CO3 saturado e o solvente foi removido sob vácuo, produzindo um óleo avermelhado pesando 3,3 g. Este foi destilado a 111-115 °C a 0,5 mm/Hg para dar um sólido verde pálido. Após recristalização a partir de benzeno, obtiveram-se 2,8 g de 1-(2,5-dimetoxi-4-metilfenil)-2-propanona sob a forma de cristais brancos com uma temperatura de 57-59 °C. Esta cetona foi também descrita como um óleo amarelo-pálido com um bp de 115-118 °C a 0,4 mm/Hg. Uma solução de 0,7 g de 1-(2,5-dimetoxifenil-4-metil)-2-propanona em 20 mL de MeOH foi tratada com 6,0 g de acetato de amónio, 0,3 g de cianoborohidreto de sódio e 3 g de peneiras moleculares Linde 3 A. A mistura foi agitada durante a noite, os sólidos foram removidos por filtração e o filtrado foi dissolvido em 100 mL de H2O. A solução foi acidificada com H2SO4 diluído e lavada com 2x25 mL de CH2Cl2. A fase aquosa foi tornada básica com NaOH aquoso, e o produto extraído com 2x25 mL de CH2Cl2. O solvente foi removido sob vácuo e o resíduo destilado (a 160 °C a 0,2 mm/Hg) para dar um produto incolor que foi dissolvido em 3 mL de IPA, neutralizado com HCl concentrado e diluído com 50 mL de Et2O anidro. Obteve-se 0,18 g de cloridrato de 2,5-dimetoxi-4-metilanfetamina (DOM) como um sólido branco com uma temperatura de 187-188 °C.
Os isómeros ópticos do DOM foram preparados de duas formas. A base racémica foi resolvida como sal de ácido orto-nitrotartranílico por recristalização a partir de EtOH. O ácido (+) fornece preferencialmente o isómero (+) ou "S" de DOM. Além disso, a 1-(2,5-dimetoxi-4-metilfenil)-2-propanona acima referida pode ser aminada redutoramente com alfa-metilbenzilamina opticamente ativa com níquel Raney. Esta amina é isolada e purificada por recristalização do sal de cloridrato. Quando opticamente pura, o grupo benzilo foi removido por hidrogenólise com paládio sobre carbono. A temperatura máxima de qualquer dos isómeros ópticos, como sais de cloridrato, é de 204-205 °C.
DOSAGEM: 3 - 10 mg.
DURAÇÃO: 14 - 20 h.
MDA
3,4-METILENODIOXIANFETAMINA
SÍNTESE: (a partir do piperonal) A uma solução de 15,0 g de piperonal em 80 mL de ácido acético glacial foram adicionados 15 mL de nitroetano, seguidos de 10 g de ciclohexilamina. A mistura foi mantida à temperatura de banho de vapor durante 6 h, diluída com 10 mL de H2O, semeada com um cristal de produto e arrefecida durante a noite a 10 °C. Os cristais amarelos brilhantes foram removidos por filtração e secos ao ar para produzir 10,7 g de 1-(3,4-metilenodioxifenil)-2-nitropropeno com um mp de 93-94 °C. Este valor foi aumentado para 97-98 °C por recristalização a partir de ácido acético. Os esforços mais convencionais de síntese do nitrostireno, utilizando um excesso de nitroetano como solvente e acetato de amónio anidro como base, dão origem a produtos impuros com rendimentos muito baixos. O nitroestireno foi obtido com sucesso a partir dos componentes em MeOH frio, com NaOH aquoso como base.
Uma suspensão de 20 g de LAH em 250 mL de THF anidro foi colocada sob uma atmosfera inerte e agitada magneticamente. Foram adicionados, gota a gota, 18 g de 1-(3,4-metilenodioxifenil)-2-nitropropeno em solução em THF e a mistura reacional foi mantida em refluxo durante 36 h. Depois de ter sido trazida de volta à temperatura ambiente, o excesso de hidreto foi destruído com 15 mL de IPA, seguido de 15 mL de NaOH a 15%. Foram adicionados mais 50 mL de H2O para completar a conversão dos sais de alumínio num sólido solto, branco e facilmente filtrável. Este foi removido por filtração, e o bolo de filtro foi lavado com THF adicional. O filtrado e as lavagens combinadas foram removidos do solvente sob vácuo e o resíduo dissolvido em H2SO4 diluído. A lavagem com 3x75 mL de CH2Cl2 removeu grande parte da cor, e a fase aquosa foi tornada básica e reextraída com 3x100 mL de CH2Cl2. A remoção do solvente produziu 13,0 g de um óleo de cor amarela que foi destilado. A fração que ferve a 80-90 °C a 0,2 mm pesou 10,2 g e era branco-água. Foi dissolvida em 60 mL de IPA, neutralizada com HCl concentrado e diluída com 120 mL de Et2O anidro, o que produziu uma turvação duradoura. Formaram-se espontaneamente cristais que foram removidos por filtração, lavados com Et2O e secos ao ar para fornecer 10,4 g de cloridrato de 3,4-metilenodioxianfetamina (MDA) com um mp de 187-188 °C.
(a partir de 3,4-metilenodioxifenilacetona) A uma solução de 32,5 g de acetato de amónio anidro em 120 mL de MeOH, foram adicionados 7,12 g de 3,4-metilenodioxifenilacetona (ver MDMA para a sua preparação), seguidos de 2,0 g de cianoborohidreto de sódio. A solução amarela resultante foi agitada vigorosamente e adicionou-se periodicamente HCl concentrado para manter o pH da mistura reacional entre 6 e 7, determinado por um papel de pH universal externo húmido. Após vários dias, os sólidos não dissolvidos permaneceram na mistura de reação e não foi necessário mais ácido. A mistura reacional foi adicionada a 600 mL de HCl diluído e esta foi lavada com 3x100 mL de CH2Cl2. As lavagens combinadas foram novamente extraídas com uma pequena quantidade de HCl diluído, as fases aquosas foram combinadas e tornadas básicas com NaOH a 25%. Em seguida, extraiu-se com 3x100 mL de CH2Cl2, estes extractos foram combinados e o solvente foi removido sob vácuo para fornecer 3,8 g de um resíduo de cor vermelha. Este foi destilado a 80-90 °C a 0,2 mm/Hg para fornecer 2,2 g de um óleo absolutamente branco-água. Não houve formação óbvia de um sal de carbonato quando exposto ao ar. Este foi dissolvido em 15 mL de IPA, neutralizado com 25 gotas de HCl concentrado, e diluído com 30 mL de Et2O anidro. Lentamente, houve a deposição de cristais brancos de cloridrato de 3,4-metilenodioxianfetamina (MDA), que pesava 2,2 g e tinha uma temperatura de 187-188 °C. A preparação da formamida (um precursor da MDMA) e da acetamida (um precursor da MDE) é descrita nessas entradas.
DOSAGEM: 80 - 160 mg.
DURAÇÃO: 4 - 6
(revisto em setembro de 2001).
MESCALINA;
3,4,5-TRIMETOXIFENETILAMINA
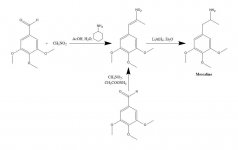
SÍNTESE: Uma solução de 20 g de 3,4,5-trimetoxibenzaldeído, 40 mL de nitrometano e 20 mL de ciclohexilamina em 200 mL de ácido acético foi aquecida no banho de vapor durante 1 h. A mistura reacional foi então diluída lentamente e com boa agitação, com 400 mL de H2O, o que permitiu a formação de uma massa cristalina amarela pesada. Esta foi removida por filtração, lavada com H2O e aspirada o mais secamente possível. A recristalização a partir de MeOH em ebulição (15 mL/g) produziu, após filtração e secagem ao ar, beta-nitro-3,4,5-trimetoxiestireno como cristais amarelos brilhantes pesando 18,5 g. Uma síntese alternativa foi eficaz, utilizando um excesso de nitrometano como solvente, bem como reagente, se a quantidade de catálise de acetato de amónio foi mantida pequena. Uma solução de 20 g de 3,4,5-trimetoxibenzaldeído em 40 mL de nitrometano contendo 1 g de acetato de amónio anidro foi aquecida no banho de vapor durante 4 h. O solvente foi removido sob vácuo e o óleo amarelo residual foi dissolvido em dois volumes de MeOH quente, decantado de alguns insolúveis e deixado arrefecer. Os cristais formados são removidos por filtração, lavados com MeOH e secos ao ar, obtendo-se 14,2 g de cristais amarelos brilhantes de beta-nitro-3,4,5-trimetoxiestireno. A utilização destas proporções, mas com 3,5 g de acetato de amónio, deu origem a extensos produtos de reação lateral, mesmo quando trabalhados após apenas 1,5 h de aquecimento. O rendimento do nitroestireno foi, neste último caso, insatisfatório.
A uma suspensão em refluxo suave de 2 g de LAH em 200 mL de Et2O, foram adicionados 2,4 g de beta-nitro-3,4,5-trimetoxiestireno como uma solução saturada de Et2O por utilização de um condensador de extração de Soxhlet modificado para permitir o retorno contínuo do solvente condensado através do dedal. Após a adição estar completa, as condições de refluxo foram mantidas durante mais 48 h. Depois de arrefecer a mistura reacional, adicionou-se cautelosamente um total de 150 mL de H2SO4 1,5 N, destruindo o excesso de hidreto e fornecendo finalmente duas fases claras. Estas foram separadas, e a fase aquosa foi lavada uma vez com 50 mL de Et2O. Adicionou-se então 50 g de tartarato de sódio e potássio, seguido de NaOH suficiente para obter um pH >9. Esta fase foi então extraída com 3x75 mL de CH2Cl2, e o solvente dos extractos agrupados foi removido sob vácuo. O resíduo foi destilado a 120-130 °C a 0,3 mm/Hg dando um óleo branco que foi dissolvido em 10 mL de IPA e neutralizado com HCl concentrado. Os cristais brancos que se formaram foram diluídos com 25 mL de Et2O, removidos por filtração e secos ao ar para fornecer 2,1 g de cloridrato de 3,4,5-trimetoxifenetilamina (M) como cristais brancos brilhantes. O sal de sulfato formou cristais espectaculares a partir da água, mas tinha um mp amplo e incaraterístico. Uma síntese alternativa pode utilizar 3,4,5-trimetoxifenilacetonitrilo, como descrito em beta-D.
DOSAGEM: 200-400 mg (como o sal de sulfato), 178-356 mg (como o sal de cloridrato)
[Nota da Erowid: O texto original dizia "178-256", mas isso foi um erro. O erro foi encontrado por Bo e verificado com Shulgin. Ver a página Erowid Mescaline Dosage para uma discussão mais completa sobre as formas de mescalina].
DURAÇÃO: 10-12 h
TMA
3,4,5-TRIMETOXIANFETAMINA
SÍNTESE: A uma solução de 39,2 g de 3,4,5-trimetoxibenzaldeído em 30 mL de EtOH morno foram adicionados 15,7 g de nitroetano seguido de 1,5 mL de n-butilamina. A mistura de reação foi deixada em repouso a 40 °C durante 7 dias. Com arrefecimento e raspagem, obtiveram-se agulhas amarelas finas que, após remoção por filtração e secagem ao ar, pesaram 48 g. A recristalização a partir de EtOH deu 2-nitro-1-(3,4,5-trimetoxifenil)propeno como cristais amarelos com um mp de 94-95 °C. Anal. (C12H15NO5) C,H,N. Em alternativa, uma solução de 20 g do aldeído em 75 mL de nitroetano foi tratada com 4 g de acetato de amónio anidro e aquecida no banho de vapor até se obter uma cor vermelha profunda. A remoção do excesso de solvente/reagente sob vácuo deu origem a um óleo vermelho que foi dissolvido num volume igual de MeOH em ebulição. Ao arrefecer, separaram-se cristais amarelos de nitropropeno. A recristalização a partir de MeOH deu, após secagem ao ar até peso constante, 13,0 g com o mesmo pH.
Sob uma atmosfera inerte, 38 g de LAH foram molhados com 100 mL de Et2O anidro, e depois suspensos em 1 L de THF seco. Este foi levado a um refluxo suave, e foi adicionado, lentamente, uma solução de 43,7 g de 2-nitro-1-(3,4,5-trimetoxifenil)propeno em 160 mL de THF. O refluxo foi continuado durante 36 h e, em seguida, a mistura reacional foi arrefecida com um banho de gelo externo. O excesso de hidreto foi destruído pela adição cautelosa de 38 mL de H2O, seguido de 38 mL de NaOH a 15% e, finalmente, mais 114 mL de H2O. Os sais inorgânicos, que deveriam ter terminado como uma massa solta, granular e facilmente filtrável, pareciam mais uma pasta de biblioteca, mas foram filtrados mesmo assim. A lavagem com THF foi tentada, mas não foi eficiente. O filtrado combinado e as lavagens foram removidos do solvente sob vácuo, dando 31,5 g da base bruta como um óleo âmbar. Este foi dissolvido em 140 mL de IPA, neutralizado com HCl concentrado (foram necessários 15 mL) e diluído com 650 mL de Et2O anidro. Houve uma fase oleosa inicial que, com agitação contínua, mudou para sólidos rosa pálido. Estes foram finamente moídos sob CH3CN para dar 15,2 g de cloridrato de 3,4,5-trimetoxianfetamina (TMA) como cristais brancos que derreteram a 195-211 °C. Todos os sais de alumínio de todo o lado foram dissolvidos em HCl diluído e foi adicionado 1 kg de tartarato de sódio e potássio. Adicionou-se NaOH a 25%, o que permitiu elevar o pH a >9 sem a precipitação de alumina básica. A extração desta fase com CH2Cl2, seguida da remoção do solvente e da formação do sal, como descrito acima, permitiu o isolamento de mais 6,4 g de TMA. O produto assim preparado contém cerca de 10-15% de 3,5-dimetoxi-4-hidroxianfetamina como impureza. Uma solução de 20 g de TMA preparada desta forma em 200 mL de NaOH a 5% foi extraída com 2x200 mL de CH2Cl2. Os extractos agrupados foram lavados com 4x100 mL de NaOH a 5% e as lavagens aquosas foram agrupadas com a fase de base original. A fase orgânica foi removida do CH2Cl2 sob vácuo para dar um óleo que foi dissolvido em 40 mL de IPA, neutralizado com HCl concentrado e diluído com 400 mL de Et2O anidro. Verificou-se a formação imediata de cristais brancos espectaculares de cloridrato de 3,4,5-trimetoxianfetamina puro, pesando 15,4 g e com uma temperatura de 220-221 °C. A fase aquosa foi levada à neutralidade, tratada com 10 g de di-hidrogenofosfato de potássio, levada a pH 9,0 com a adição cuidadosa de NaOH, e extraída com 5x100 mL de CH2Cl2. A evaporação do solvente sob vácuo deu origem a um óleo que cristalizou espontaneamente. Este produto, 3,5-dimetoxi-4-hidroxianfetamina, pode ser purificado por
sublimação a 130 °C a 0,2 mm/Hg. Trata-se de um sólido cristalino branco que descolora lentamente ao ar. A literatura descreve um sal picrato com uma temperatura de 225 °C a partir de EtOH.
DOSAGEM: 100 - 250 mg.
DURAÇÃO: 6 - 8 h.