2C-B
4-BROM-2,5-DIMETOXI-FENETILAMIN
SZINÓZIS: 100 g 2,5-dimetoxi-benzaldehid 220 g nitrometánban lévő oldatát 10 g vízmentes ammónium-acetáttal kezeltük, és gőzfürdőn 2,5 órán át melegítettük, időnként kevergetve. A mélyvörös reakcióelegyből vákuumban eltávolítottuk a felesleges nitrometánt, és a maradék spontán kikristályosodott. Ezt a nyers nitrosztirént IPA alatt történő őrléssel, szűréssel és légszárítással tisztítottuk, így 85 g 2,5-dimetoxi-béta-nitrosztirént kaptunk, amely sárgás-narancs színű, a következő lépéshez megfelelő tisztaságú termék volt. További tisztítás forró IPA-ból történő átkristályosítással érhető el.
Egy mágneskeverővel ellátott és inert atmoszféra alá helyezett, kerek aljú 2 L-es lombikba 750 ml vízmentes THF-et adtunk, amely 30 g LAH-t tartalmazott. Ezután THF-oldatban 60 g 2,5-dimetoxi-béta-nitrosztirént adtunk hozzá. A kész oldat piszkos sárgásbarna színű volt, és 24 órán keresztül reflux hőmérsékleten tartottuk. Lehűtés után a felesleges hidridet IPA cseppenkénti hozzáadásával megsemmisítettük. Ezután 30 ml 15%-os NaOH-t adtunk hozzá, hogy a szervetlen szilárd anyagot szűrhető masszává alakítsuk. A reakcióelegyet leszűrtük, és a szűrőpogácsát először THF-fel, majd MeOH-val mostuk. Az egyesített anyalúgokat és mosófolyadékokat vákuumban mentesítettük az oldószertől, és a maradékot 1,5 l H2O-ban szuszpendáltuk. Ezt HCl-lel savasítottuk, 3x100 ml CH2Cl2-vel mostuk, 25%-os NaOH-val erősen bázissá tettük, és 4x100 ml CH2Cl2-vel újra extraháltuk. Az egyesített kivonatokból vákuumban eltávolítottuk az oldószert, így 26 g olajos maradékot kaptunk, amelyet 120-130 °C-on, 0,5 mm/Hg nyomáson desztilláltunk, így 21 g fehér olajat, 2,5-dimetoxi-fenetilamint
(2C-H) kaptunk, amely a levegőből nagyon gyorsan felveszi a szén-dioxidot.
A 24,8 g 2,5-dimetoxi-fenetil-amin 40 ml jégecetben jól kevert oldatához 22 g elemi brómot adtunk 40 ml ecetsavban oldva. Néhány perc múlva szilárd anyag képződése és egyidejűleg jelentős hőfejlődés következett be. A reakcióelegyet visszaengedtük szobahőmérsékletre, leszűrtük, és a szilárd anyagot hideg ecetsavval kíméletesen kimostuk. Ez volt a hidrobrómsó. Számos bonyolult sóforma létezik, mind polimorfok, mind hidrátok, amelyek alattomossá tehetik a 2C-B izolálását és jellemzését. A legszerencsésebb út az oldhatatlan hidroklorid só képzése a szabad bázis útján. Az ecetsavval nedvesített só teljes tömegét meleg H2O-ban feloldottuk, 25%-os NaOH-val legalább pH 11-re bázissá tettük, és 3x100 ml CH2Cl2-vel extraháltuk. Az oldószer eltávolítása 33,7 g maradékot eredményezett, amelyet 115-130 °C-on, 0,4 mm/Hg nyomáson desztilláltunk. A 27,6 g fehér olajat feloldottuk 50 ml H2O-ban, amely 7,0 g ecetsavat tartalmazott. Ezt a tiszta oldatot erőteljesen kevertettük, majd 20 mL tömény HCl-lel kezeltük. Azonnal képződött a 2,5-dimetoxi-4-bromfenetil-amin-hidroklorid vízmentes sója (2C-B). Ezt a kristálytömeget szűréssel eltávolítottuk (további 60 mL H2O hozzáadásával jelentősen fellazítható), kevés H2O-val, majd több 50 mL Et2O-val mostuk. Teljesen légszáraz állapotban 31,05 g finom fehér tűt kaptunk, amelynek mp értéke 237-239 °C volt bomlással. Ha a végső tömény HCl hozzáadásakor túl sok H2O van jelen, a 2C-B hidratált formáját kapjuk. A hidrobrómsó 214,5-215 °C-on olvad. Az acetát sóról azt jelentették, hogy az mp 208-209 °C.
ADAGOLÁS: 12-24 mg.
HATÓSÁG: 4-8 óra.
DOM
STP; 2,5-DIMETOXI-4-METIL-AMFETAMIN
SZINTHEZIS: 54,9 g 2,5-dimetoxi-4-metil-benzaldehid (előállítását lásd a
2C-D receptben) 215 g jégecetsavban lévő oldatához 19,5 g vízmentes ammónium-acetátot és 30,6 g nitroetánt adtunk. Ezt az elegyet 3 órán keresztül gőzfürdőn melegítettük, a reakcióelegyet nedves jégfürdőn hűtöttük, lehetővé téve a sárga kristályok spontán képződését. Annyi H2O-t adtunk hozzá, amennyit csak tudtunk (éppen csak a tartósan zavaros olajos jelleget nem elérve), és további néhány óra állás után a kristályos 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropént szűréssel eltávolítottuk, és forrásban lévő ecetsavból átkristályosítottuk. Az állandó tömegre való szárítás után a hozam 28,3 g volt, az mp 87-88 °C volt. Anal. (C12H15NO4) C, H ,N.
9,5 g LAH szuszpenzióját 750 ml jól kevert vízmentes Et2O-ban inert atmoszféra alatt refluxon tartottuk, a kondenzált oldószert egy 9,5 g 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropént tartalmazó Soxhlet gyűszűn keresztül visszavezetve. Miután a nitrosztirén hozzáadása befejeződött, a kevert szuszpenziót további 4 órán keresztül refluxon tartottuk, majd szobahőmérsékletre hűtöttük, és egy éjszakán át hagytuk tovább keverni. A felesleges hidridet 750 mL 8%-os H2SO4 hozzáadásával semmisítettük meg, óvatosan, amíg a hidrogénfejlődés meg nem szűnt, majd olyan sebességgel, hogy a képződött szilárd anyag diszpergálódjon. A fázisokat elválasztottuk, a vizes fázist egyszer Et2O-val mostuk, 225 g kálium-nátrium-tartaráttal kezeltük, végül 5%-os NaOH-val bázissá (pH >9) tettük. Ezt 3x150 mL CH2Cl2-vel extraháltuk, a kivonásokat összevontuk, és az oldószert vákuumban eltávolítottuk. A maradék 9,6 g tiszta olaj volt, amely hexánból spontán kristályokat képzett 60,5-61 °C-os mp-értékkel. Ezeket a szilárd anyagokat 150 ml vízmentes Et2O-ban feloldottuk, és vízmentes HCl-gázzal telítettük. Miután 2 órán át szobahőmérsékleten állt, a kristályos 2,5-dimetoxi-4-metil-amfetamin-hidrokloridot (DOM) szűréssel eltávolítottuk, Et2O-val mostuk, és levegőn súlyállandóságig szárítottuk. 8,25 g csillogó fehér kristályokat kaptunk, amelyek MP értéke 190,5-191,5 °C volt. A szulfát mp értéke 131 °C volt. Analitika. (C12H20ClNO2) C, H ,N.
A fenti nitrosztirén a megfelelő fenilaceton közvetítésével is átalakítható a végtermékké. Visszafolyási hőmérsékleten tartott 20 ml jégecetsavban 10,4 g vaspor jól kevert szuszpenziójához 4,9 g 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropént adtunk szilárd anyagként. A refluxálást 2 órán keresztül folytattuk, majd az egészet nedves Celiten keresztül szűrtük. 300 ml H2O-val, majd 300 ml Et2O-val történő mosás után az egyesített szűrletet és a mosásokat elválasztottuk, és a vizes fázist 2x100 ml Et2O-val extraháltuk. A szerves fázist és az extraktumokat egyesítettük, majd 2x100 mL telített K2CO3-mal mostuk, és az oldószert vákuumban eltávolítottuk, így 3,3 g vöröses színű olajat kaptunk. 111-115 °C-on, 0,5 mm/Hg nyomáson desztilláltuk, és halványzöld szilárd anyagot kaptunk. Benzolból történő átkristályosítás után 2,8 g 1-(2,5-dimetoxi-4-metilfenil)-2-propanont kaptunk, fehér kristályok formájában, 57-59 °C közötti hőmérsékleten. Ezt a keton halványsárga olajként is leírták, amelynek bp értéke 115-118 °C 0,4 mm/Hg mellett. 0,7 g 1-(2,5-dimetoxi-fenil-4-metil)-2-propanon 20 ml MeOH-ban lévő oldatát 6,0 g ammónium-acetáttal, 0,3 g nátrium-cianoborohidriddel és 3 g Linde 3 A molekulaszitával kezeltük. Az elegyet egy éjszakán át kevertettük, a szilárd anyagokat szűréssel eltávolítottuk, és a szűrletet 100 mL H2O-ban feloldottuk. Az oldatot híg H2SO4-mal savasítottuk, majd 2x25 mL CH2Cl2-vel mostuk. A vizes fázist vizes NaOH-val bázissá tettük, és a terméket 2x25 mL CH2Cl2-vel extraháltuk. Az oldószert vákuumban eltávolítottuk, és a maradékot desztilláltuk (160 °C-on, 0,2 mm/Hg mellett), hogy színtelen terméket kapjunk, amelyet 3 mL IPA-ban feloldottunk, tömény HCl-lel semlegesítettünk, és 50 mL vízmentes Et2O-val hígítottunk. Így 0,18 g 2,5-dimetoxi-4-metil-amfetamin-hidrokloridot (DOM) kaptunk fehér szilárd anyagként, amelynek mp értéke 187-188 °C volt.
A DOM optikai izomerjeit kétféle módon állítottuk elő. A racém bázist EtOH-ból történő átkristályosítással oldottuk fel orto-nitrotartranilsav sóként. A (+) sav a DOM (+) vagy "S" izomerjét adja előszeretettel. A fent említett 1-(2,5-dimetoxi-4-metilfenil)-2-propanon is reduktívan aminálható optikailag aktív alfa-metil-benzilaminnal Raney Nickel segítségével. Ezt az amint a hidroklorid só átkristályosításával izolálják és tisztítják. Amikor optikailag tiszta, a benzilcsoportot palládium-szénnel végzett hidrogenolízissel távolították el. A hidroklorid sóként kapott optikai izomerek bármelyikének mp értéke 204-205 °C volt.
ADAGOLÁS: 3-10 mg.
HATÓSÁG: 14-20 óra.
MDA
3,4-METILÉN-DIOXIAMFETAMIN
SZINÓZIS: (piperonálból) 15,0 g piperonál 80 ml jégecetsavban lévő oldatához 15 ml nitroetánt, majd 10 g ciklohexilamint adtunk. Az elegyet 6 órán keresztül gőzfürdő hőmérsékleten tartottuk, 10 mL H2O-val hígítottuk, a termék kristályával beoltottuk, és egy éjszakán át 10 °C-on hűtöttük. Az élénksárga kristályokat szűréssel eltávolítottuk, majd levegőn szárítottuk, így 10,7 g 1-(3,4-metiléndioxiilfenil)-2-nitropropént kaptunk, amelynek mp értéke 93-94 °C volt. Ezt ecetsavból történő átkristályosítással 97-98 °C-ra emeltük. A nitrosztirénszintézis hagyományosabb erőfeszítései, amelyek során többlet nitroetánt használnak oldószerként és vízmentes ammónium-acetátot bázisként, nagyon gyenge hozamú, tisztátalan terméket adnak. A nitrosztirént sikeresen állították elő a komponensekből hideg MeOH-ban, vizes NaOH-val mint bázissal.
20 g LAH szuszpenzióját 250 ml vízmentes THF-ben inert atmoszféra alá helyeztük és mágnesesen kevertettük. Cseppenként 18 g 1-(3,4-metiléndioxifenil)-2-nitropropil THF-ben oldott 18 g 1-(3,4-metiléndioxifenil)-2-nitropropilént adtunk hozzá, és a reakcióelegyet 36 órán át refluxon tartottuk. 15 mL IPA-val, majd 15 mL 15%-os NaOH-val a szobahőmérsékletre való visszahozás után a felesleges hidridet megsemmisítettük. További 50 mL H2O-t adtunk hozzá, hogy az alumíniumsók átalakulása laza, fehér, könnyen szűrhető szilárd anyaggá fejeződjön be. Ezt szűréssel távolítottuk el, és a szűrőpogácsát további THF-fel mostuk. Az egyesített szűrletet és a mosásokat vákuumban eltávolítottuk az oldószertől, és a maradékot híg H2SO4-ban feloldottuk. A 3x75 mL CH2Cl2-vel történő mosás eltávolította a szín nagy részét, a vizes fázist bázissá tettük, és 3x100 mL CH2Cl2-vel újra extraháltuk. Az oldószer eltávolításával 13,0 g sárga színű olajat nyertünk, amelyet desztilláltunk. A 80-90 °C-on 0,2 mm-en forrásban lévő frakció 10,2 g súlyú és vízfehér volt. Feloldottuk 60 mL IPA-ban, tömény HCl-lel semlegesítettük, majd 120 mL vízmentes Et2O-val hígítottuk, ami tartós zavarosságot eredményezett. Spontán kristályok képződtek, amelyeket szűréssel eltávolítottunk, Et2O-val mostunk és levegőn szárítottuk, így 10,4 g 3,4-metilén-dioxiamfetamin-hidrokloridot (MDA) kaptunk, amelynek mp értéke 187-188 °C volt.
(3,4-metiléndioxi-fenilacetonból) 32,5 g vízmentes ammónium-acetát 120 ml MeOH-ban lévő oldatához 7,12 g 3,4-metiléndioxi-fenilacetont (előállítását lásd az MDMA-nál), majd 2,0 g nátrium-cianoborohidridet adtunk. Az így kapott sárga oldatot erőteljesen kevertettük, és időnként tömény HCl-t adtunk hozzá, hogy a reakcióelegy pH-ját 6 és 7 között tartsuk, ahogyan azt külső nedves univerzális pH-papírral határoztuk meg. Néhány nap elteltével a reakcióelegyben oldatlan szilárd anyagok maradtak, és nem volt szükség több savra. A reakcióelegyet 600 mL híg HCl-hez adtuk, és ezt 3x100 mL CH2Cl2-vel mostuk. Az egyesített mosásokat kis mennyiségű híg HCl-lel visszanyertük, a vizes fázisokat egyesítettük, és 25%-os NaOH-val bázissá tettük. Ezt követően 3x100 ml CH2Cl2-vel extraháltuk, ezeket a kivonásokat egyesítettük, és az oldószert vákuumban eltávolítottuk, így 3,8 g vörös színű maradékot kaptunk. Ezt 80-90 °C-on, 0,2 mm/Hg nyomáson desztilláltuk, így 2,2 g teljesen vízfehér olajat kaptunk. Levegőn nem képződött nyilvánvalóan karbonátos só. Ezt 15 ml IPA-ban feloldottuk, 25 csepp tömény HCl-lel semlegesítettük, és 30 ml vízmentes Et2O-val hígítottuk. Lassan lerakódtak a 3,4-metiléndioxiamfetamin-hidroklorid (MDA) fehér kristályai, amelyek tömege 2,2 g volt, mp-je 187-188 °C. A kristályok a következő: 3,4-metiléndioxiamfetamin-hidroklorid (MDA). A formamid (az MDMA prekurzora) és az acetamid (az MDE prekurzora) előállítását a fenti bejegyzések alatt ismertetjük.
ADAGOLÁS: 80-160 mg.
IDŐTARTAM: 4 - 6
(felülvizsgálva, 2001. szeptember).
MESZKÁLIN;
3,4,5-TRIMETOXI-FENETILAMIN
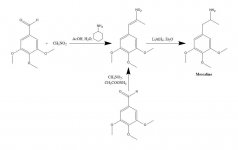
SZINTÉZIS: 20 g 3,4,5-trimetoxi-benzaldehid, 40 ml nitrometán és 20 ml ciklohexil-amin 200 ml ecetsavban lévő oldatát gőzfürdőn 1 órán át melegítettük, majd a reakcióelegyet lassan és jó keverés mellett 400 ml H2O-val hígítottuk, ami lehetővé tette egy nehéz, sárga kristályos tömeg képződését. Ezt szűréssel eltávolítottuk, H2O-val mostuk, és a lehető legszárazabban leszívtuk. Forrásban lévő MeOH-ból (15 ml/g) történő átkristályosítás után szűrés és levegőn történő szárítás után a béta-nitro-3,4,5-trimetoxisztirolt kaptuk élénksárga kristályokként, 18,5 g tömegben. Egy másik szintézis is hatékony volt, amelynél a nitrometán feleslegét használtuk oldószerként és reagensként is, ha az ammónium-acetát katalizátor mennyiségét kicsiben tartottuk. 20 g 3,4,5-trimetoxi-benzaldehid 40 ml nitrometánban lévő, 1 g vízmentes ammónium-acetátot tartalmazó oldatát gőzfürdőn 4 órán át melegítettük. Az oldószert vákuumban lecsupaszítottuk, és a visszamaradt sárga olajat két térfogat forró MeOH-ban feloldottuk, néhány oldatlanként lefejtettük, és hagytuk kihűlni. A képződött kristályokat szűréssel eltávolítottuk, MeOH-val mostuk és levegőn szárítottuk, így 14,2 g béta-nitro-3,4,5-trimetoxisztirol élénksárga kristályokat kaptunk. Ezeknek az arányoknak a használata, de 3,5 g ammónium-acetát hozzáadásával, még 1,5 óra hevítés után is kiterjedt mellékreakciós termékeket adott. A nitrosztirén hozama ez utóbbi esetben nem volt kielégítő.
200 ml Et2O-ban 2 g LAH gyengén visszafolyó szuszpenziójához 2,4 g béta-nitro-3,4,5-trimetoxisztirolt adtunk telített Et2O-oldatként, egy Soxhlet extrakciós kondenzátor segítségével, amelyet úgy módosítottunk, hogy a kondenzált oldószer folyamatosan visszajusson a gyűszűre. Miután az addíció befejeződött, a refluxálási körülményeket további 48 órán keresztül fenntartottuk. A reakcióelegy lehűtése után összesen 150 ml 1,5 N H2SO4-ot adtunk hozzá óvatosan, elpusztítva a felesleges hidridet, és végül két tiszta fázist biztosítva. Ezeket elválasztottuk, és a vizes fázist egyszer 50 mL Et2O-val mostuk. Ezután 50 g kálium-nátrium-tartarátot adtunk hozzá, majd annyi NaOH-t, hogy a pH >9 legyen. Ezt követően 3x75 mL CH2Cl2-vel extraháltuk, majd az egyesített extraktumokból az oldószert vákuumban eltávolítottuk. A maradékot 120-130 °C-on, 0,3 mm/Hg nyomáson desztilláltuk, és fehér olajat kaptunk, amelyet 10 mL IPA-ban feloldottunk és tömény HCl-lel semlegesítettünk. A képződött fehér kristályokat 25 ml Et2O-val hígítottuk, szűréssel eltávolítottuk és levegőn szárítottuk, így 2,1 g 3,4,5-trimetoxi-fenetilamin-hidrokloridot (M) kaptunk csillogó fehér kristályokként. A szulfát só vízből látványos kristályokat képzett, de széles és jellegtelen mp-vel rendelkezett. Alternatív szintézisben a béta-D pontban leírtak szerint 3,4,5-trimetoxi-fenilacetonitrilt használhatunk.
ADAGOLÁS: 200-400 mg (mint szulfát só), 178-356 mg (mint hidroklorid só)
[Erowid megjegyzés: Az eredeti szövegben "178-256" szerepelt, de ez hiba volt. A hibát Bo találta meg és ellenőrizte Shulginnal. A
meszkalin formáinak részletesebb tárgyalását lásd az
Erowid Mescaline Dosage oldalán].
Időtartam: 10-12 óra
TMA
3,4,5-TRIMETOXI-AMFETAMIN
SZINTÉZIS: 39,2 g 3,4,5-trimetoxi-benzaldehid 30 ml meleg EtOH-ban lévő oldatához 15,7 g nitroetánt, majd 1,5 ml n-butil-amint adtunk. A reakcióelegyet 40 °C-on 7 napig hagytuk állni. Lehűtéssel és kaparással finom sárga tűket kaptunk, amelyek szűréssel és levegőn való szárítással történő eltávolítás után 48 g súlyúak voltak. EtOH-ból történő átkristályosítással a 2-nitro-1-(3,4,5-trimetoxifenil)propén sárga kristályok formájában 94-95 °C-os hőmérsékletű volt. Anal. (C12H15NO5) C,H,N. Alternatív megoldásként 20 g aldehid 75 ml nitroetánban lévő oldatát 4 g vízmentes ammónium-acetáttal kezeltük és gőzfürdőn addig hevítettük, amíg mélyvörös színt nem kaptunk. A felesleges oldószer/reagens vákuumban történő eltávolítása vörös olajat eredményezett, amelyet feloldottunk ugyanolyan térfogatú forrásban lévő MeOH-ban. Lehűtéskor a nitropropilén sárga kristályai váltak szét. MeOH-ból történő átkristályosítás után levegőn tömegállandóságig történő szárítás után 13,0 g-ot kaptunk, ugyanolyan mp-vel.
Inert atmoszféra alatt 38 g LAH-t 100 ml vízmentes Et2O-val nedvesítettünk, majd 1 L száraz THF-ben szuszpendáltuk. Ezt enyhe visszaáramlásig hoztuk, majd lassan hozzáadtuk 43,7 g 2-nitro-1-(3,4,5-trimetoxifenil)propén oldatát 160 mL THF-ben. A refluxálást 36 órán keresztül folytattuk, majd a reakcióelegyet külső jégfürdővel hűtöttük. A felesleges hidridet 38 mL H2O óvatos hozzáadásával semmisítettük meg, majd ezt 38 mL 15%-os NaOH, végül újabb 114 mL H2O követte. A szervetlen sók, amelyeknek laza, szemcsés, könnyen szűrhető masszaként kellett volna végbemenniük, inkább könyvtári pasztának tűntek, de ennek ellenére leszűrtük őket. Megkíséreltük a mosást THF-fel, de nem volt hatékony. Az egyesített szűrletből és a mosásokból vákuumban eltávolítottuk az oldószert, így 31,5 g nyers bázist kaptunk borostyánszínű olajként. Ezt 140 ml IPA-ban feloldottuk, tömény HCl-lel semlegesítettük (15 ml-re volt szükség), és 650 ml vízmentes Et2O-val hígítottuk. Kezdetben olajos fázis keletkezett, amely folyamatos keverés hatására halvány rózsaszínű szilárd anyaggá változott. Ezeket CH3CN alatt finomra őröltük, így 15,2 g 3,4,5-trimetoxi-amfetamin-hidrokloridot (TMA) kaptunk fehér kristályokként, amelyek 195-211 °C-on olvadtak. Az összes, mindenhonnan származó alumíniumsót híg HCl-ben feloldottuk, és 1 kg kálium-nátrium-tartarátot adtunk hozzá. Ott, mint hozzáadott 25% NaOH lehetővé tette a pH >9-re történő csökkentését a bázikus timföld kicsapódása nélkül. E fázis CH2Cl2-vel történő extrakcióját az oldószer eltávolítása és a sóképződés követte a fent leírtak szerint, ami további 6,4 g TMA izolálását tette lehetővé. Az így előállított termék mintegy 10-15% 3,5-dimetoxi-4-hidroxi-amfetamint tartalmaz szennyeződésként. Az így előállított 20 g TMA 200 ml 5%-os NaOH-ban oldott 20 g oldatát 2x200 ml CH2Cl2-vel extraháltuk. Az egyesített extraktumokat 4x100 ml 5%-os NaOH-val mostuk, és a vizes mosásokat az eredeti bázisfázissal egyesítettük. A szerves fázist vákuumban lecsupaszítottuk a CH2Cl2-től, így olajat kaptunk, amelyet 40 mL IPA-ban feloldottunk, tömény HCl-lel semlegesítettünk, és 400 mL vízmentes Et2O-val hígítottunk. Azonnal látványos, fehér színű, tiszta 3,4,5-trimetoxi-amfetamin-hidroklorid kristályok keletkeztek, amelyek tömege 15,4 g volt és 220-221 °C-os mp-értékkel rendelkeztek. A vizes fázist semleges állapotba hoztuk, 10 g kálium-di-hidrogén-foszfáttal kezeltük, NaOH óvatos hozzáadásával pH 9,0-ra hoztuk, és 5x100 ml CH2Cl2-vel extraháltuk. Az oldószer vákuumban történő elpárologtatásával olaj keletkezett, amely spontán kristályosodott. Ez a termék, a 3,5-dimetoxi-4-hidroxi-amfetamin 130 °C-on, 0,2 mm/Hg nyomáson történő
szublimálással tovább tisztítható volt. Fehér kristályos szilárd anyag volt, amely a levegőn lassan elszíneződött. Az irodalomban EtOH-ból származó pikrát-sót írnak le, amelynek mp-je 225 °C volt.
ADAGOLÁS: 100-250 mg.
IDŐTARTAM: 6 - 8 óra.