
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,792
- Solutions
- 3
- Reaction score
- 3,053
- Points
- 113
- Deals
- 1
Introducción
El ácido lisérgico, el fragmento básico derivado de los alcaloides del cornezuelo de centeno, se ha sintetizado en una secuencia catorce que comienza con 3-beta-carboxietilindol. El material de partida se convirtió en el intermediario 1-benzoil-5-ceto-1,2,2a-3,4,5-hexahidrobenzo-[cd]-indol (3), que contiene tres de los cuatro anillos presentes en el ácido lisérgico. Esta cetona se transformó a su vez en el compuesto tetracíclico, 9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4.3-fe]-quinolina (8), y de ahí en ácido l isérgico (14). Esta síntesis no es sencilla y requiere mucha experiencia de laboratorio y conocimientos de química. Además, hay varias manipulaciones con sustancias peligrosas, que deben llevarse a cabo con estrictas medidas de seguridad.
Punto de ebullición: 536,2±50,0 °C a 760 mm Hg;
Punto de fusión: 240 °C;
Peso molecular: 268,31 g/ mol;
Densidad: 1,4 ± 0,1 g/mL;
Número CAS: 82-58-6.
Equipo y cristalería:
- Reactor de hidrogenación de acero 2-3 L;
- Autoclave de acero 500 mL;
- Balanza de laboratorio (adecuada de 0,01 a 500 g);
- Matraces de fondo redondo de 100, 200, 500 mL, 5 y 10 L;
- Compresor de hidrógeno (H2) y origen;
- Matraz Buchner y embudo (grande) de 5 L [puede utilizarse un filtro Schott para pequeñas cantidades];
- Máquina Rotovap (grande);
- Fuente de vacío;
- Embudos de separación de 500 mL y 2 L;
- Globo de nitrógeno ~50-70 L (1 bar);
- Tapones de septo para matraces;
- Baño de agua helada con sal;
- 5 L x2, 2 L x2; 1 L x2; 500 mL x2; 100 mL x3; 50 mL x2 Vasos de precipitados;
- Jeringa de vidrio o pipeta Pasteur;
- Agitador magnético o agitador superior;
- Instalación de destilación al vacío;
- Condensador de reflujo;
- Soporte de retorta y abrazadera para fijar el aparato;
- Termómetro de laboratorio (de -20 °C a 200 °C) con adaptador para matraz;
- Papel indicador del pH;
- Varilla de vidrio y espátula;
- Bombilla de 250 vatios.
Reactivos.
- Ácido 3-indolpropiónico (1), 94,6 g (0,5 mol);
- 9,5 L Agua destilada (H2O);
- ~400 g de hidróxido de sodio (NaOH);
- 116 g de níquel Raney (Ni);
- 1050 mL Ácido clorhídrico (HCl) concentrado;
- 2 mL de ácido sulfúrico (H2SO4 conc.);
- 210 mL de solución 12N de hidróxido sódico (NaOH) aq;
- 180 mL Cloruro de benzoilo;
- ~1,5 L Metanol (MeOH);
- ~1,6 L de etanol (EtOH);
- 201,2 mL Cloruro de tionilo (SOCl2);
- 1950 mL Disulfuro de carbono (CS2);
- 240 g de cloruro de aluminio (AlCl3);
- 2,5 L de benceno;
- 500 mL Hidróxido sódico 2N (NaOH);
- ~3,2 L Éter dietílico (Et2O);
- 3,3 L Ácido acético glacial (AcOH);
- 352 g (1,1 mol) Hidrobromuro de piridina perbromuro;
- 5 L Cloroformo (CHCl3);
- ~1000 g Sulfato de magnesio (MgSO4);
- 307 g (2,35 mol) Metilaminoacetona etileno cetal (5);
- 4,5 L Benceno;
- ~500 g Carbón activado (C);
- ~1 L Acetona;
- ~500 g Bicarbonato sódico (NaHCO3);
- 80 mL Anhídrido acético frío (Ac2O);
- 1,5 g Borohidruro sódico (NaBH4);
- 75 mL Dióxido de azufre (SO2 líquido);
- 40 g de cianuro sódico (NaCN polvo);
- 300 mL Cianuro de hidrógeno (HCN líquido);
- 78 mL Solución acuosa de hidróxido de potasio (KOH) al 1,5
- 8,5 g Arseniato sódico hidratado;
- ~ 50 mL Xileno;
- 100 mL Solución diluida de hidróxido de amonio (NH4OH);
- 16,9 g de metóxido de sodio (MeONa).
Procedimiento
1-Benzoil-3-(beta-carboxietil)-2,3-dihidroindol (2)El ácido 3-indolpropiónico (1), 94,6 g (0,5 mol), se disolvió en 600 mL de agua que contenía 20 g de hidróxido sódico. La solución se mezcló con unos 100 g de catalizador de níquel Raney y se hidrogenó a TA en una bomba de hidrogenación de acero de 2-3 L a 3000-4000 psi (207-276 bar) de presión H2. La reducción solía completarse en 20-30 h, tras lo cual se filtraba el catalizador y se lavaba con un poco de agua. Se añadía ácido HCl concentrado, 85 mL, al filtrado y se enfriaba la solución. Si la reducción era incompleta, el ácido indolpropiónico sin reaccionar se separaba en este punto y se eliminaba por filtración. A continuación, el filtrado se benzoiló por el procedimiento habitual de Schotten-Baumann, utilizando 210 mL de hidróxido sódico 12N y 180 mL de cloruro de benzoilo. La solución se mantuvo alcalina durante toda la benzoilación y la temperatura se mantuvo por debajo de 40 °C mediante enfriamiento. Cuando el cloruro de benzoilo reaccionó completamente, la mezcla se enfrió y se acidificó con 300 mL de ácido HCl concentrado. El producto bruto se filtró y se lavó con agua, tras lo cual se extrajo con 4 porciones de 1 L de agua caliente para eliminar el ácido benzoico. El producto almibarado caliente (2), tras decantación del extracto acuoso, se cristalizó a partir de unos pocos volúmenes de metanol; rendimiento 103 g (70 %), MP: 151-153 °C.
1-Benzoil-5-ceto-1,2,2a,3,4,5-hexahidrobenzo-[cd]-indol (3)
1-Benzoil-3-(beta-carboxietil)-2,3-dihidroindol (2), 118 g (0,4 mol), se mezcló con 200 mL de cloruro de tionilo puro. La disolución se dejó reposar durante 30 min, después de lo cual se calentó suavemente durante 15-20 min en el baño de vapor. El exceso de cloruro de tionilo se evaporó completamente al vacío por debajo de 30 °C y el cloruro ácido bruto se disolvió en 200 mL de disulfuro de carbono. La disolución del cloruro ácido se añadió entonces en chorro fino a una suspensión vigorosamente agitada de 240 g de cloruro de aluminio en 1750 mL de disulfuro de carbono contenida en un matraz de 5 L (¡¡¡en HORNO!!!). Se separó un complejo y la agitación se hizo difícil. La mezcla se calentó a reflujo y se agitó durante una hora para completar la reacción, tras lo cual se descompuso con mucho cuidado añadiendo 500 g de hielo, 250 mL de ácido HCl conc. y 500 mL de agua. Durante la descomposición, se mantuvo la agitación y el enfriamiento se afectó por destilación periódica del disulfuro de carbono in vacuo, y el producto se extrajo con 2 L de benceno. El extracto se lavó a fondo con 500 mL de hidróxido sódico 2N en tres porciones y después con agua. Se secó sobre sulfato de magnesio y se evaporó al vacío hasta un volumen pequeño. La adición lenta de varios volúmenes de éter hizo cristalizar la cetona amarilla (3) . Se filtró y se lavó con éter; rendimiento 85,3 g (77 %), MP: 146-147 °C. Se recristalizó una muestra para su análisis a partir de benceno-éter.
1-Benzoil-3-(beta-carboxietil)-2,3-dihidroindol (2), 118 g (0,4 mol), se mezcló con 200 mL de cloruro de tionilo puro. La disolución se dejó reposar durante 30 min, después de lo cual se calentó suavemente durante 15-20 min en el baño de vapor. El exceso de cloruro de tionilo se evaporó completamente al vacío por debajo de 30 °C y el cloruro ácido bruto se disolvió en 200 mL de disulfuro de carbono. La disolución del cloruro ácido se añadió entonces en chorro fino a una suspensión vigorosamente agitada de 240 g de cloruro de aluminio en 1750 mL de disulfuro de carbono contenida en un matraz de 5 L (¡¡¡en HORNO!!!). Se separó un complejo y la agitación se hizo difícil. La mezcla se calentó a reflujo y se agitó durante una hora para completar la reacción, tras lo cual se descompuso con mucho cuidado añadiendo 500 g de hielo, 250 mL de ácido HCl conc. y 500 mL de agua. Durante la descomposición, se mantuvo la agitación y el enfriamiento se afectó por destilación periódica del disulfuro de carbono in vacuo, y el producto se extrajo con 2 L de benceno. El extracto se lavó a fondo con 500 mL de hidróxido sódico 2N en tres porciones y después con agua. Se secó sobre sulfato de magnesio y se evaporó al vacío hasta un volumen pequeño. La adición lenta de varios volúmenes de éter hizo cristalizar la cetona amarilla (3) . Se filtró y se lavó con éter; rendimiento 85,3 g (77 %), MP: 146-147 °C. Se recristalizó una muestra para su análisis a partir de benceno-éter.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
1-Benzoil-2,2a,3,4-tetrahidro-4-[metil-(2-metil-1,2-dioxolan-2-il-metil)-amino]-benz-[cd]-indol-5-(1H)-ona (6)
Una solución de 270 g (0,76 mol) de 1-benzoil-4-bromo-5-ceto-1,2,2a,3,4,5-hexahidrobenz-[cd]-indol(4) y 307 g (2.35 mol) de metilaminoacetona etileno cetal (5) en 4500 mL de benceno seco se sometió a reflujo bajo nitrógeno durante 21 h en RBF de 10 L con condensador de reflujo. La mezcla se enfrió, y se filtraron 151 g (93,5 %) de hidrobromuro de metilaminoacetona etileno cetal, MP: 158-159 °C.
El filtrado se lavó varias veces con agua helada, después de lo cual se extrajo con 2,5 L de ácido HCl diluido frío conteniendo 150 mL del ácido concentrado. Los extractos ácidos se añadieron inmediatamente a un exceso de hidróxido sódico diluido frío con hielo. El producto se extrajo con 1 L de cloroformo, y la disolución de cloroformo se secó sobre sulfato de magnesio, se trató con carbón y se concentró al vacío. Elcetal-cetona residual (6) se cristalizó a partir de acetona; MP: y MP de la mezcla: 135-136 °C, rendimiento 220 g (71 %).
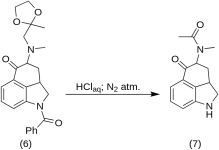
5-Ceto-4-[N-metil-N-acetonilamino]-1,2,2a,3,4,5-hexahidrobenz-[cd]-indol (7)
20 g de 1-benzoil-2,2a,3,4-tetrahidro-4-[metil-(2-metil-1,3-dioxolan-2-il-metil)-amino]-benz-[cd]-indol-5-(1H)-ona (6) se disolvió en una mezcla de 250 mL de ácido HCl concentrado y 250 mL de agua, y la solución se mantuvo bajo nitrógeno a 37 °C en RBF de 3-5 L durante cinco días. La mezcla se enfrió, se trató con carbón, se filtró y el filtrado se concentró in vacuo a pequeño volumen. El residuo se trató con bicarbonato sódico en exceso; el producto se extrajo con cloroformo frío y el disolvente se eliminó al vacío a temperatura ambiente. La diketona bruta (7) se pulverizó, se mezcló con unos 75 mL de benceno-éter 1:1 y se filtró; rendimiento 9,8 g (77 %), MP: 105-107 °C. Una muestra para análisis se recristalizó en una botella de vidrio. Serecristalizó una muestra para análisis a partir de benceno-éter o etanol; MP: 109-110 °C; se obtuvo un monohidrocloruro a partir de etanol diluido; MP: 200 °C dec.
9-Keto-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (8)
Se mezclaron 25 g de 5-Keto-4-[N-metil-N-acetonil]-amino-1,2,2a,3,4,5-hexahidrobenz-[cd]-indol (7) con 550 mL de etanol absoluto. La mezcla se agitó bajo nitrógeno y se enfrió a -15 °C en 2-5 L de RBF. A continuación se añadió metóxido sódico, 16,9 g, y la mezcla se agitó a -10 °C a -12 °C durante diez minutos. La mezcla de reacción se enfrió a -25 °C, y el producto se filtró en un embudo Buchner de 6,5 pulgadas y se lavó con un poco de etanol frío y éter. Con la mínima exposición al aire (¡contiene metóxido sódico!), la cetona bruta (8) se tamizó inmediatamente con un poco de agua helada y se volvió a filtrar. Se lavó con agua helada, etanol y éter; rendimiento 16,2 g (69 %), MP: 145-147 °C. Una muestra analítica se recristalizó con agua helada. Una muestra analítica se recristalizó a partir de etanol diluido; MP: 155-157 °C; El dihidrocloruro se preparó y recristalizó a partir de acetona acuosa; MP: 270 °C dec.
4-Acetil-9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina(9)
Se añadió 9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (8), 24 g, a 80 mL de anhídrido acético frío. La mezcla se mantuvo a 25 °C en 200 mL de RBF durante unos 5 min, tras lo cual se enfrió completamente, y el producto (9) se filtró y se lavó con éter; rendimiento 20,5 g (76 %), mp: 167-170 °C. Se obtuvo una segunda cosecha por evaporación del filtrado; esto elevó el rendimiento total al 82 %. Una muestra se recristalizó de acetona-etanol; MP: 169-170 °C; El clorhidrato se preparó en etanol y se recristalizó de etanol acuoso; MP: 250 °C dec.
Se añadió 9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (8), 24 g, a 80 mL de anhídrido acético frío. La mezcla se mantuvo a 25 °C en 200 mL de RBF durante unos 5 min, tras lo cual se enfrió completamente, y el producto (9) se filtró y se lavó con éter; rendimiento 20,5 g (76 %), mp: 167-170 °C. Se obtuvo una segunda cosecha por evaporación del filtrado; esto elevó el rendimiento total al 82 %. Una muestra se recristalizó de acetona-etanol; MP: 169-170 °C; El clorhidrato se preparó en etanol y se recristalizó de etanol acuoso; MP: 250 °C dec.
4-Acetil-9-hidroxi-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (10)
10 g de 4-acetil-9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octahidroxindolo-[4,3-fg]-quinolina ( 9) a una mezcla de 150 mL de metanol y 10 mL de agua en 500 mL de RBF. Se añadió borohidruro sódico, 1,5 g, y se dejó que la reacción avanzara a RT hasta un volumen pequeño, y se añadió una mezcla de 15 mL de ácido HCl conc. y 60 mL de agua. El clorhidrato (10) que se separó al enfriarse se filtró y se lavó con metanol, 9,0 g (79 %). Se recristalizó una muestra a partir de etanol diluido; MP: 245-246 °C dec.
Clorhidrato de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina(11)
El clorhidrato de 4-acetil-9-hidroxi-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (10), 3,1 g, se disolvió en 75 ml de agua.1 g, se disolvió en 75 mL de dióxido de azufre líquido contenido en una camisa de vidrio en un autoclave de acero de 500 mL. Se añadió cloruro de tionilo, 1,2 mL, y el recipiente se selló y se mantuvo a 25 °C durante 6 h. Se purgó el autoclave y se extrajo la mezcla de reacción. Se dejó evaporar el dióxido de azufre mientras se mantenía constante el volumen de la disolución mediante la adición lenta de éter seco. El clorhidrato amorfo (11) se filtró, se lavó con éter y se secó al vacío, MP: 130-135 °C dec. Rendimiento 3,5 g. Eluso del alcohol 9-beta-epimérico en esta reacción dio el mismo cloruro en rendimiento comparable.
4-Acetil-9-ciano-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (12)
Se añadieron 40 g de cianuro sódico seco en polvo a 300 mL de cianuro de hidrógeno líquido helado. La mezcla se agitó y se enfrió en hielo, y se añadieron 7,5 g del hidrocloruro de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo [4,3f/g]-quinolina (11 ) bruto amorfo anterior. Se continuó agitando en 500 mL de RBF durante 30 min, después de lo cual el cianuro de hidrógeno se destiló rápidamente a presión reducida por debajo de unos 10 °C. El residuo se mezcló con cloroformo y agua helada, y la mezcla resultante se filtró. La capa orgánica se separó y la fase acuosa se extrajo dos veces con cloroformo. Los extractos combinados se secaron sobre sulfato de magnesio, se decoloraron y el disolvente se destiló al vacío. El producto (12) se cristalizó a partir de acetato de etilo; rendimiento 3,3 g. (54% sobre la base del clorhidrato de alcohol), p.m. 172-174 °C. Larecristalización a partir del mismo disolvente elevó la p.m. a 181-182 °C.
Se añadieron 40 g de cianuro sódico seco en polvo a 300 mL de cianuro de hidrógeno líquido helado. La mezcla se agitó y se enfrió en hielo, y se añadieron 7,5 g del hidrocloruro de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo [4,3f/g]-quinolina (11 ) bruto amorfo anterior. Se continuó agitando en 500 mL de RBF durante 30 min, después de lo cual el cianuro de hidrógeno se destiló rápidamente a presión reducida por debajo de unos 10 °C. El residuo se mezcló con cloroformo y agua helada, y la mezcla resultante se filtró. La capa orgánica se separó y la fase acuosa se extrajo dos veces con cloroformo. Los extractos combinados se secaron sobre sulfato de magnesio, se decoloraron y el disolvente se destiló al vacío. El producto (12) se cristalizó a partir de acetato de etilo; rendimiento 3,3 g. (54% sobre la base del clorhidrato de alcohol), p.m. 172-174 °C. Larecristalización a partir del mismo disolvente elevó la p.m. a 181-182 °C.
9-Carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (13)
El producto (12) anterior, 1,0 g, se mezcló con 15 mL de metanol y 0,25 mL de agua. La mezcla se enfrió y se añadieron lentamente 2 mL de ácido sulfúrico concentrado. La solución se selló en un tubo de vidrio bajo nitrógeno y se calentó a 100 °C durante 23 a 24 h en RBF de 100 mL con condensador de reflujo. La mezcla se trató con carbón decolorado y después se concentró al vacío hasta unos 10 mL. Se vertió sobre una mezcla de cloroformo (30 mL), hielo y 10 g de bicarbonato sódico. Se separó la capa de cloroformo y la fase acuosa se extrajo con 3 porciones de 10 mL de cloroformo. Los extractos combinados se secaron sobre sulfato de magnesio, se evaporaron hasta sequedad y el producto (13) se cristalizó a partir de benceno; rendimiento 0,51 g (53 %), MP: 159-160 °C. Se recristalizó a partir de etanol. Se recristalizó a partir de acetato de etilo; MP: 160-161 °C.
El producto (12) anterior, 1,0 g, se mezcló con 15 mL de metanol y 0,25 mL de agua. La mezcla se enfrió y se añadieron lentamente 2 mL de ácido sulfúrico concentrado. La solución se selló en un tubo de vidrio bajo nitrógeno y se calentó a 100 °C durante 23 a 24 h en RBF de 100 mL con condensador de reflujo. La mezcla se trató con carbón decolorado y después se concentró al vacío hasta unos 10 mL. Se vertió sobre una mezcla de cloroformo (30 mL), hielo y 10 g de bicarbonato sódico. Se separó la capa de cloroformo y la fase acuosa se extrajo con 3 porciones de 10 mL de cloroformo. Los extractos combinados se secaron sobre sulfato de magnesio, se evaporaron hasta sequedad y el producto (13) se cristalizó a partir de benceno; rendimiento 0,51 g (53 %), MP: 159-160 °C. Se recristalizó a partir de etanol. Se recristalizó a partir de acetato de etilo; MP: 160-161 °C.
Ácido dl-lisérgico sintético (14)
Una mezcla de 9-carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (13), 3,9 g, y 78 mL de solución de hidróxido potásico al 1,5 % se sometió a reflujo durante 30 min bajo nitrógeno. Se añadió arseniato sódico hidratado, 8,5 g, y níquel Raney (16 g, húmedo), previamente desactivado por ebullición en suspensión de xileno, y la mezcla se calentó a reflujo y se agitó en atmósfera de nitrógeno durante 20 horas en 200 mL de RBF con condensador de reflujo. La solución se trató con carbón, y el ácido lisérgico bruto (14) se precipitó por neutralización a pH 5,6. Se filtró y se lavó con ácido lisérgico. Se filtró y se lavó con agua; rendimiento 1,04 g, MP: 240-242 °C dec. También se obtuvo una segunda cosecha, 0,16 g, MP: 233-235 °C dec.; rendimiento total 30%. El ácido pudo purificarse disolviéndolo en hidróxido de amonio diluido, tratándolo con carbón decolorante y reprecipitándolo con dióxido de carbono., MP: 242-243 °C dec; una mezcla con ácido dl-lisérgico hecha a partir de ácido d-lisérgico natural fue igualmente de 242-243 °C dec. El ácido anhidro se obtuvo secando al vacío durante varias horas a 150 °C.
Una mezcla de 9-carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (13), 3,9 g, y 78 mL de solución de hidróxido potásico al 1,5 % se sometió a reflujo durante 30 min bajo nitrógeno. Se añadió arseniato sódico hidratado, 8,5 g, y níquel Raney (16 g, húmedo), previamente desactivado por ebullición en suspensión de xileno, y la mezcla se calentó a reflujo y se agitó en atmósfera de nitrógeno durante 20 horas en 200 mL de RBF con condensador de reflujo. La solución se trató con carbón, y el ácido lisérgico bruto (14) se precipitó por neutralización a pH 5,6. Se filtró y se lavó con ácido lisérgico. Se filtró y se lavó con agua; rendimiento 1,04 g, MP: 240-242 °C dec. También se obtuvo una segunda cosecha, 0,16 g, MP: 233-235 °C dec.; rendimiento total 30%. El ácido pudo purificarse disolviéndolo en hidróxido de amonio diluido, tratándolo con carbón decolorante y reprecipitándolo con dióxido de carbono., MP: 242-243 °C dec; una mezcla con ácido dl-lisérgico hecha a partir de ácido d-lisérgico natural fue igualmente de 242-243 °C dec. El ácido anhidro se obtuvo secando al vacío durante varias horas a 150 °C.
Attachments

Last edited:
