2C-B
4-БРОМО-2,5-ДИМЕТОКСИФЕНЕТИЛАМИН
Синтез: Разтвор на 100 g 2,5-диметоксибензалдехид в 220 g нитрометан се третира с 10 g безводен амониев ацетат и се нагрява на парна баня в продължение на 2,5 h при периодично завъртане. Тъмночервената реакционна смес се отстранява от излишния нитрометан под вакуум и остатъкът кристализира спонтанно. Този суров нитростирен се пречиства чрез смилане под IPA, филтриране и сушене на въздух, за да се получат 85 g 2,5-диметокси-бета-нитростирен като жълто-оранжев продукт с достатъчна чистота за следващата стъпка. Допълнително пречистване може да се постигне чрез рекристализация от кипящ IPA.
В кръглодънна колба от 2 L, снабдена с магнитна бъркалка и поставена под инертна атмосфера, се добавят 750 ml безводен THF, съдържащ 30 g LAH. След това в разтвор на THF се добавят 60 g 2,5-диметокси-бета-нитростирен. Окончателният разтвор е с мръсно жълто-кафяв цвят и се държи при температура на рефлукс в продължение на 24 h. След охлаждане излишният хидрид се унищожава чрез капково добавяне на IPA. След това се добавят 30 ml 15% NaOH, за да се превърнат неорганичните твърди вещества във филтрируема маса. Реакционната смес се филтрира, а филтърният кек се промива първо с THF, а след това с MeOH. Комбинираните изходни течности и промивки се освобождават от разтворителя под вакуум и остатъкът се суспендира в 1,5 L H2O. Тя се подкиселява с HCl, промива се с 3x100 mL CH2Cl2, прави се силно основна с 25% NaOH и се екстрахира отново с 4x100 mL CH2Cl2. Събраните екстракти се лишават от разтворителя под вакуум, като се получава 26 g маслена утайка, която се дестилира при 120-130 °C при 0,5 mm/Hg, за да се получи 21 g бяло масло, 2,5-диметокси-фенетиламин
(2C-H), което много бързо поема въглероден диоксид от въздуха.
Към добре разбъркан разтвор на 24,8 g 2,5-диметоксифенетиламин в 40 ml ледена оцетна киселина се добавят 22 g елементарен бром, разтворен в 40 ml оцетна киселина. След няколко минути се наблюдава образуване на твърди частици и едновременно с това отделяне на значително количество топлина. Реакционната смес се оставя да се върне на стайна температура, филтрира се и твърдите частици се промиват пестеливо със студена оцетна киселина. Това е хидробромидната сол. Съществуват много сложни солни форми, както полиморфи, така и хидрати, които могат да направят изолирането и характеризирането на 2С-В коварно. Най-щастливият път е да се образува неразтворимата хидрохлоридна сол чрез свободна основа. Цялата маса на намокрената с оцетна киселина сол се разтваря в топла H2O, прави се основна до най-малко pH 11 с 25 % NaOH и се екстрахира с 3x100 ml CH2Cl2. След отстраняване на разтворителя се получава 33,7 g остатък, който се дестилира при 115-130 °C и 0,4 mm/Hg. Бялото масло, 27,6 g, се разтваря в 50 mL H2O, съдържащо 7,0 g оцетна киселина. Този бистър разтвор се разбърква енергично и се обработва с 20 ml концентрирана HCl. Незабавно се образува безводна сол на 2,5-диметокси-4-бромофенетиламин хидрохлорид (2С-В). Тази маса от кристали се отстранява чрез филтриране (тя може да се разхлаби значително чрез добавяне на още 60 mL H2O), промива се с малко H2O, а след това с няколко 50 mL порции Et2O. При пълно изсушаване на въздуха се получиха 31,05 g фини бели иглички с mp 237-239 °C с разлагане. Когато в момента на добавяне на последната концентрирана HCl има твърде много H2O, се получава хидратирана форма на 2С-В. Хидробромидната сол се топи при 214,5-215 °C. Съобщава се, че ацетатната сол има mp от 208-209 °C.
ДОЗИРОВКА: 12 - 24 mg.
ПРОДЪЛЖИТЕЛНОСТ: 4 - 8 часа.
DOM
STP; 2,5-ДИМЕТОКСИ-4-МЕТИЛАМФЕТАМИН
СИНТЕЗА: Към разтвор на 54,9 g 2,5-диметокси-4-метилбензалдехид (за приготвянето му виж рецептата за
2С-D ) в 215 g ледена оцетна киселина са добавени 19,5 g безводен амониев ацетат и 30,6 g нитроетан. Тази смес се нагрява в продължение на 3 h на парна баня, реакционната смес се охлажда в мокра ледена баня, което позволява спонтанното образуване на жълти кристали. Добавя се възможно най-голямо количество H2O (малко преди да се получи устойчив мътен маслен характер) и след още няколко часа престой кристалният 1-(2,5-диметокси-4-метилфенил)-2-нитропропен се отстранява чрез филтриране и се рекристализира от кипяща оцетна киселина. Добивът, след изсушаване до постоянно тегло, е 28,3 g, а mp е 87-88 °C. Анализ. (C12H15NO4) C, H, N.
Суспензия от 9,5 g LAH в 750 ml добре разбъркан безводен Et2O се държи при обратен поток в инертна атмосфера, като връщането на кондензирания разтворител става през накрайник на Сокслет, съдържащ 9,5 g 1-(2,5-диметокси-4-метилфенил)-2-нитропропен. След като добавянето на нитростирена е завършило, разбърканата суспензия е поддържана при рефлукс за още 4 часа, след което е охладена до стайна температура и е оставена да продължи да се разбърква през нощта. Излишъкът от хидрид се унищожава чрез добавяне на 750 ml 8% H2SO4, внимателно, докато се прекрати отделянето на водород, след което със скорост, която позволява на образуваните твърди вещества да се диспергират. Фазите се разделят, водната фаза се промива веднъж с Et2O, обработва се с 225 g калиев натриев тартарат и накрая се прави основна (pH >9) с 5% NaOH. Това се екстрахира с 3х150 ml CH2Cl2, екстрактите се събират и разтворителят се отстранява под вакуум. Остатъкът е 9,6 g прозрачно масло, което спонтанно образува кристали с mp от 60,5-61 °C от хексан. Тези твърди частици се разтварят в 150 ml безводен Et2O и се насищат с безводен газ HCl. След престой на стайна температура в продължение на 2 h кристалният 2,5-диметокси-4-метиламфетаминов хидрохлорид (DOM) се отстранява чрез филтриране, промива се с Et2O и се изсушава на въздух до постоянно тегло. Получиха се 8,25 g блестящи бели кристали с mp от 190,5-191,5 °C. Сулфатът има mp от 131 °C. Анализ. (C12H20ClNO2) C, H, N.
Горепосоченият нитростирен може също да се превърне в крайния аминов продукт чрез междинния продукт на съответния фенилацетон. Към добре разбъркана суспензия от 10,4 g желязо на прах в 20 ml ледена оцетна киселина, държана при температура на обратен поток, се добавят 4,9 g 1-(2,5-диметокси-4-метилфенил)-2-нитропропен като твърдо вещество. Охлаждането е продължило 2 часа, след което всичко е филтрирано през мокър целит. След промиване с 300 mL H2O, последвано от 300 mL Et2O, комбинираният филтрат и промивките са отделени, а водната фаза е екстрахирана с 2x100 mL Et2O. Органичната фаза и екстрактите се комбинират и се промиват с 2x100 mL наситен K2CO3 и разтворителят се отстранява под вакуум, като се получава червеникаво масло с тегло 3,3 g. То се дестилира при 111-115 °C при 0,5 mm/Hg, за да се получи бледозелено твърдо вещество. След рекристализация от бензол се получават 2,8 g 1-(2,5-диметокси-4-метилфенил)-2-пропанон като бели кристали с mp 57-59 °C. Този кетон е описан и като бледожълто масло с bp от 115-118 °C при 0,4 mm/Hg. Разтвор на 0,7 g 1-(2,5-диметоксифенил-4-метил)-2-пропанон в 20 ml MeOH се третира с 6,0 g амониев ацетат, 0,3 g натриев цианоборохидрид и 3 g молекулни сита Linde 3 A. Сместа се разбърква за една нощ, твърдите частици се отстраняват чрез филтриране, а филтратът се разтваря в 100 ml H2O. Разтворът се подкиселява с разреден H2SO4 и се промива с 2x25 mL CH2Cl2. Водната фаза се прави основна с воден NaOH, а продуктът се екстрахира с 2x25 mL CH2Cl2. Разтворителят е отстранен под вакуум, а остатъкът е дестилиран (при 160 °C при 0,2 mm/Hg), за да се получи безцветен продукт, който е разтворен в 3 mL IPA, неутрализиран с концентриран HCl и разреден с 50 mL безводен Et2O. Получават се 0,18 g 2,5-диметокси-4-метиламфетамин хидрохлорид (DOM) като бяло твърдо вещество с mp 187-188 °C.
Оптичните изомери на DOM са получени по два начина. Рацемичната основа е била разтворена като сол на орто-нитротраниловата киселина чрез рекристализация от EtOH. (+) киселината осигурява (+) или "S" изомера на DOM с предимство. Също така гореспоменатият 1-(2,5-диметокси-4-метилфенил)-2-пропанон може да бъде редуктивно аминиран с оптично активен алфа-метилбензиламин с никел на Раней. Този амин се изолира и пречиства чрез рекристализация на хидрохлоридната сол. Когато е оптически чист, бензиловата група се отстранява чрез хидрогенолиза с паладий върху въглерод. Мр на всеки от оптичните изомери, като хидрохлоридни соли, е 204-205 °C.
ДОЗИРОВКА: 3 - 10 mg.
ПРОДЪЛЖИТЕЛНОСТ: 14 - 20 часа.
MDA
3,4-МЕТИЛЕНДИОКСИАМФЕТАМИН
СИНТЕЗ: (от пиперонал) Към разтвор на 15,0 g пиперонал в 80 ml ледена оцетна киселина се добавят 15 ml нитроетан, последван от 10 g циклохексиламин. Сместа се държи при температура на парна баня в продължение на 6 h, разрежда се с 10 mL H2O, посява се с кристал от продукта и се охлажда за една нощ при 10 °C. Яркожълтите кристали се отстраняват чрез филтриране и се изсушават на въздух, за да се получат 10,7 g 1-(3,4-метилендиоксифенил)-2-нитропропен с mp 93-94 °C. То се повишава до 97-98 °C чрез рекристализация от оцетна киселина. По-конвенционалните опити за синтез на нитростирен, при които се използва излишък от нитроетан като разтворител и безводен амониев ацетат като основа, дават нечист продукт с много ниски добиви. Нитростиренът е получен успешно от компонентите в студен MeOH, с воден NaOH като основа.
Суспензия от 20 g LAH в 250 ml безводен THF се поставя под инертна атмосфера и се разбърква магнитно. Добавят се по капки 18 g 1-(3,4-метилендиоксифенил)-2-нитропропен в разтвор в THF и реакционната смес се поддържа при обратен поток в продължение на 36 h. След като се върне на стайна температура, излишният хидрид се унищожава с 15 mL IPA, последван от 15 mL 15% NaOH. Добавени са още 50 mL H2O, за да се завърши превръщането на алуминиевите соли в насипно, бяло, лесно филтрируемо твърдо вещество. То се отстранява чрез филтриране, а филтърният кек се промива допълнително с THF. Комбинираният филтрат и промивките се лишават от разтворителя под вакуум, а остатъкът се разтваря в разредена H2SO4. Промиването с 3х75 mL CH2Cl2 премахва голяма част от цвета, а водната фаза се прави основна и се екстрахира отново с 3х100 mL CH2Cl2. След отстраняване на разтворителя се получава 13,0 g жълто оцветено масло, което се дестилира. Фракцията, кипяща при 80-90 °С при 0,2 mm, е с тегло 10,2 g и е воднобяла. Тя се разтваря в 60 ml IPA, неутрализира се с концентрирана HCl и се разрежда със 120 ml безводен Et2O, при което се получава трайно помътняване. Спонтанно се образуват кристали, които се отстраняват чрез филтриране, промиват се с Et2O и се изсушават на въздух, за да се получи 10,4 g 3,4-метилендиоксиамфетамин хидрохлорид (MDA) с mp 187-188 °C.
(от 3,4-метилендиоксифенилацетон) Към разтвора на 32,5 g безводен амониев ацетат в 120 ml MeOH се добавят 7,12 g 3,4-метилендиоксифенилацетон (за получаването му вижте в раздела MDMA), последван от 2,0 g натриев цианоборохидрид. Полученият жълт разтвор се разбърква енергично и периодично се добавя концентриран HCl, за да се поддържа рН на реакционната смес между 6 и 7, както е определено чрез външна влажна универсална рН хартия. След няколко дни в реакционната смес остават неразтворени твърди вещества и не е необходима повече киселина. Реакционната смес е добавена към 600 mL разредена HCl и е промита с 3x100 mL CH2Cl2. Комбинираните промивки се екстрахират обратно с малко количество разреден HCl, водните фази се комбинират и се базират с 25% NaOH. След това се екстрахира с 3x100 mL CH2Cl2, тези екстракти се комбинират и разтворителят се отстранява под вакуум, за да се получи 3,8 g червен остатък. Той се дестилира при 80-90 °С при 0,2 mm/Hg, за да се получи 2,2 g абсолютно водно бяло масло. Не се наблюдава очевидно образуване на карбонатна сол при излагане на въздух. То се разтваря в 15 ml IPA, неутрализира се с 25 капки концентриран HCl и се разрежда с 30 ml безводен Et2O. Бавно се отлагат бели кристали на 3,4-метилендиоксиамфетамин хидрохлорид (MDA), които тежат 2,2 g и имат mp 187-188 °C. Приготвянето на формамид (прекурсор на MDMA) и ацетамид (прекурсор на MDE) е описано в тези записи.
ДОЗИРОВКА: 80-160 mg.
ПРОДЪЛЖИТЕЛНОСТ: 4 - 6
(ревизирана, септември 2001 г.).
МЕСКАЛИН;
3,4,5-ТРИМЕТОКСИФЕНЕТИЛАМИН
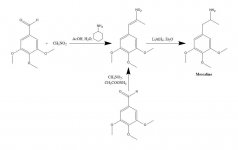
СИНТЕЗА: Разтвор на 20 g 3,4,5-триметоксибензалдехид, 40 ml нитрометан и 20 ml циклохексиламин в 200 ml оцетна киселина се нагрява на парна баня в продължение на 1 h. След това реакционната смес се разрежда бавно и при добро разбъркване с 400 ml H2O, което позволява образуването на тежка жълта кристална маса. Тя се отстранява чрез филтриране, промива се с H2O и се изсмуква възможно най-суха. При рекристализация от кипящ MeOH (15 mL/g) след филтриране и сушене на въздух се получи бета-нитро-3,4,5-триметоксистирен като яркожълти кристали с тегло 18,5 g. Алтернативен синтез е ефективен, като се използва излишък от нитрометан като разтворител, както и като реагент, ако количеството на катализа на амониев ацетат се запази малко. Разтвор на 20 g 3,4,5-триметоксибензалдехид в 40 ml нитрометан, съдържащ 1 g безводен амониев ацетат, се нагрява на парна баня в продължение на 4 h. Разтворителят се отстранява под вакуум, а остатъчното жълто масло се разтваря в два обема горещ MeOH, декантира се от някои неразтворими частици и се оставя да се охлади. Образуваните кристали се отстраняват чрез филтриране, промиват се с MeOH и се изсушават на въздух, като се получават 14,2 g яркожълти кристали на бета-нитро-3,4,5-триметоксистирен. При използването на тези пропорции, но с 3,5 g амониев ацетат, се получават обширни продукти на странични реакции, дори когато се изработват само след 1,5 h нагряване. Добивът на нитростирен в този последен случай беше незадоволителен.
Към леко рефлуксираща суспензия от 2 g LAH в 200 ml Et2O са добавени 2,4 g бета-нитро-3,4,5-trimethoxystyrene като наситен разтвор на Et2O чрез използване на екстракционен кондензатор на Сокслет, модифициран така, че да позволява непрекъснато връщане на кондензирания разтворител през напръстник. След като добавянето е завършено, условията на рефлуксиране се поддържат за още 48 h. След охлаждане на реакционната смес внимателно се добавят общо 150 mL 1,5 N H2SO4, като се унищожава излишният хидрид и в крайна сметка се получават две чисти фази. Те се разделят, а водната фаза се промива веднъж с 50 mL Et2O. След това се добавят 50 g калиев натриев тартарат, последван от достатъчно NaOH, за да се достигне pH >9. След това се екстрахира с 3х75 mL CH2Cl2 и разтворителят от събраните екстракти се отстранява под вакуум. Остатъкът се дестилира при 120-130 °C при 0,3 mm/Hg, като се получава бяло масло, което се разтваря в 10 ml IPA и се неутрализира с концентрирана HCl. Образувалите се бели кристали се разреждат с 25 ml Et2O, отстраняват се чрез филтриране и се изсушават на въздух, за да се получат 2,1 g 3,4,5-триметоксифенетиламин хидрохлорид (М) като блестящи бели кристали. Сулфатната сол образува ефектни кристали от вода, но има широк и нехарактерен mp. При алтернативен синтез може да се използва 3,4,5-триметоксифенилацетонитрил, както е описано в точка бета-Д.
ДОЗИРОВКА: 200-400 mg (като сулфатна сол), 178-356 mg (като хидрохлоридна сол)
[Забележка на Erowid: Оригиналният текст гласи "178-256", но това е грешка. Грешката е открита от Бо и е проверена при Шулгин. За по-пълно обсъждане на формите на мескалина вижте страницата на Erowid "Дозировка на мескалина" ].
ПРОДЪЛЖИТЕЛНОСТ: 10-12 часа
TMA
3,4,5-ТРИМЕТОКСИАМФЕТАМИН
Синтез: Към разтвор на 39,2 g 3,4,5-триметоксибензалдехид в 30 mL топъл EtOH се добавят 15,7 g нитроетан, последван от 1,5 mL n-бутиламин. Реакционната смес е оставена да престои при 40 °C в продължение на 7 дни. При охлаждане и надраскване се получават фини жълти иглички, които след отстраняване чрез филтриране и изсушаване на въздуха тежат 48 g. При рекристализация от EtOH се получава 2-нитро-1-(3,4,5-триметоксифенил)пропен като жълти кристали с mp 94-95 °C. Анализ. (C12H15NO5) C,H,N. Алтернативно, разтвор на 20 g от алдехида в 75 ml нитроетан се третира с 4 g безводен амониев ацетат и се нагрява на парна баня до получаване на тъмночервен цвят. Отстраняването на излишния разтворител/реагент под вакуум дава червено масло, което се разтваря в същия обем кипящ MeOH. При охлаждане се отделят жълти кристали на нитропропена. След рекристализация от MeOH се получава, след изсушаване на въздуха до постоянно тегло, 13,0 g със същото mp.
В инертна атмосфера 38 g LAH се навлажнява със 100 ml безводен Et2O, след което се суспендира в 1 L сух THF. Това се довежда до лек рефлукс и се добавя бавно разтвор на 43,7 g 2-нитро-1-(3,4,5-триметоксифенил)пропен в 160 mL THF. Охлаждането е продължило 36 h, след което реакционната смес е била охладена с външна ледена баня. Излишният хидрид е унищожен чрез внимателно добавяне на 38 mL H2O, последвано от 38 mL 15% NaOH и накрая още 114 mL H2O. Неорганичните соли, които би трябвало да се получат като рохкава, гранулирана, лесно филтрируема маса, приличаха по-скоро на библиотечна паста, но въпреки това бяха филтрирани. Беше направен опит за промиване с THF, но той не беше ефективен. Комбинираният филтрат и промивките бяха лишени от разтворител под вакуум, като се получи 31,5 g сурова основа под формата на кехлибарено масло. То се разтваря в 140 ml IPA, неутрализира се с концентриран HCl (необходими са 15 ml) и се разрежда с 650 ml безводен Et2O. Първоначално е имало маслена фаза, която при продължително разбъркване е преминала в бледорозово твърдо вещество. Те бяха фино смлени под CH3CN, за да се получат 15,2 g 3,4,5-триметоксиамфетамин хидрохлорид (ТМА) като бели кристали, които се топят при 195-211 °C. Всички алуминиеви соли отвсякъде се разтварят в разредена HCl и се добавя 1 kg калиев натриев тартарат. Там, както и добавеният 25 % NaOH, позволиха да се доведе рН до >9 без утаяване на основен алуминий. Екстракцията на тази фаза с CH2Cl2 , последвана от отстраняване на разтворителя и образуването на соли, както е описано по-горе, позволи изолирането на допълнителни 6,4 g TMA. Полученият по този начин продукт съдържа около 10-15 % 3,5-диметокси-4-хидроксиамфетамин като примес. Разтвор на 20 g от така приготвения ТМА в 200 ml 5% NaOH се екстрахира с 2x200 ml CH2Cl2. Събраните екстракти се промиват с 4х100 mL 5% NaOH, а водните промивки се събират с оригиналната основна фаза. Органичната фаза се отстранява от CH2Cl2 под вакуум, за да се получи масло, което се разтваря в 40 mL IPA, неутрализира се с концентрирана HCl и се разрежда с 400 mL безводен Et2O. Незабавно се образуват ефектни бели кристали от чист 3,4,5-триметоксиамфетамин хидрохлорид с тегло 15,4 g и с mp 220-221 °C. Водната фаза се довежда до неутралност, обработва се с 10 g калиев ди-хидрогенфосфат, довежда се до рН 9,0 с внимателно добавяне на NaOH и се екстрахира с 5x100 ml CH2Cl2. При изпаряване на разтворителя във вакуум се получава масло, което спонтанно кристализира. Този продукт, 3,5-диметокси-4-хидроксиамфетамин, може да бъде допълнително пречистен чрез
сублимация при 130 °С при 0,2 mm/Hg. Той представлява бяло кристално твърдо вещество, което бавно се обезцветява на въздуха. В литературата е описана пикратна сол с mp от 225 °C от EtOH.
ДОЗИРОВКА: 100 - 250 mg.
ПРОДЪЛЖИТЕЛНОСТ: 6 - 8 часа.