
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,792
- Solutions
- 3
- Reaction score
- 3,053
- Points
- 113
- Deals
- 1
Introdução
O ácido lisérgico, o fragmento básico derivado dos alcalóides da cravagem do centeio, foi sintetizado numa sequência de catorze, começando com o 3-beta-carboxietilindole. O material de partida foi convertido no intermediário 1-benzoil-5-ceto-1,2,2a-3,4,5-hexa-hidrobenz-[cd]-indole (3), que contém três dos quatro anéis presentes no ácido lisérgico. Esta cetona, por sua vez, foi transformada no composto tetracíclico 9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4.3-fe]-quinolina (8) e, em seguida, no ácido lisérgico (14). Esta síntese não é simples e requer muita experiência laboratorial e conhecimentos de química. Além disso, há várias manipulações com substâncias perigosas, que têm de ser efectuadas com medidas de segurança rigorosas.
Ponto de ebulição: 536,2±50,0 °C a 760 mm Hg;
Ponto de fusão: 240 °C;
Peso molecular: 268,31 g/mole;
Densidade: 1,4 ± 0,1 g/mL;
Número CAS: 82-58-6.
Equipamento e material de vidro:
- Reator de hidrogenação em aço de 2-3 L;
- Autoclave de aço de 500 mL;
- Balança de laboratório (0,01 - 500 g é adequada);
- Balões de fundo redondo de 100, 200, 500 mL, 5 e 10 L;
- Compressor de hidrogénio (H2) e origem;
- Balão de Buchner e funil (grande) de 5 L [pode ser utilizado um filtro Schott para pequenas quantidades];
- Máquina Rotovap (grande);
- Fonte de vácuo;
- Funis de separação de 500 mL e 2 L;
- Balão de azoto ~50-70 L (1 bar);
- Tampas de septo para os frascos;
- Banho de água gelada com sal;
- 5 L x2, 2 L x2; 1 L x2; 500 mL x2; 100 mL x3; 50 mL x2 Copos;
- Seringa de vidro ou pipeta de Pasteur;
- Agitador magnético ou agitador de topo;
- Instalação de destilação em vácuo;
- Condensador de refluxo;
- Suporte de retorta e pinça para fixar o aparelho;
- Termómetro de laboratório (-20 °C a 200 °C) com adaptador de balão;
- Papel indicador de pH;
- Vareta de vidro e espátula;
- Lâmpada de 250 watts.
Reagentes.
- Ácido 3-indolepropiónico (1), 94,6 g (0,5 mol);
- 9,5 L de água destilada (H2O);
- ~400 g de hidróxido de sódio (NaOH);
- 116 g de níquel Raney (Ni);
- 1050 mL de ácido clorídrico (HCl) concentrado;
- 2 mL de ácido sulfúrico (H2SO4 conc.);
- 210 mL de solução aq de hidróxido de sódio (NaOH) 12N;
- 180 mL de cloreto de benzoílo;
- ~1,5 L de metanol (MeOH);
- ~1,6 L de etanol (EtOH);
- 201,2 mL de cloreto de tionilo (SOCl2);
- 1950 mL Dissulfureto de carbono (CS2);
- 240 g de cloreto de alumínio (AlCl3);
- 2,5 L de benzeno;
- 500 mL de hidróxido de sódio 2N (NaOH);
- ~3,2 L de éter dietílico (Et2O);
- 3,3 L de ácido acético glacial (AcOH);
- 352 g (1,1 mol) de perbrometo de piridina;
- 5 L Clorofórmio (CHCl3);
- ~1000 g Sulfato de magnésio (MgSO4);
- 307 g (2,35 mol) Metilaminoacetona etileno-cetal (5);
- 4,5 L Benzeno;
- ~500 g de carvão ativado (C);
- ~1 L de acetona;
- ~500 g de bicarbonato de sódio (NaHCO3);
- 80 mL de anidrido acético frio (Ac2O);
- 1,5 g de borohidreto de sódio (NaBH4);
- 75 mL de dióxido de enxofre (SO2 líquido);
- 40 g de cianeto de sódio (NaCN em pó);
- 300 mL de cianeto de hidrogénio (HCN líquido);
- 78 mL de solução aquosa de hidróxido de potássio (KOH) a 1,5%;
- 8,5 g de arseniato de sódio hidratado;
- ~ 50 mL de xileno;
- 100 mL de solução diluída de hidróxido de amónio (NH4OH);
- 16,9 g de metóxido de sódio (MeONa).
Procedimento
1-Benzoil-3-(beta-carboxietil)-2,3-di-hidroindole (2)O ácido 3-indolepropiónico (1), 94,6 g (0,5 mol), foi dissolvido em 600 mL de água contendo 20 g de hidróxido de sódio. A solução foi misturada com cerca de 100 g de catalisador de níquel Raney e hidrogenada à temperatura ambiente numa bomba de hidrogenação de aço de 2-3 L a uma pressão de 3000-4000 psi (207-276 bar) H2. A redução foi geralmente completa em 20-30 h, após o que o catalisador foi filtrado e lavado com um pouco de água. Adicionou-se ácido HCl concentrado, 85 mL, ao filtrado e a solução foi arrefecida. Se a redução foi incompleta, o ácido indolepropiónico que não reagiu separou-se neste ponto e foi removido por filtração. O filtrado foi então benzoilado pelo procedimento habitual de Schotten-Baumann, utilizando 210 mL de hidróxido de sódio 12N e 180 mL de cloreto de benzoílo. A solução foi mantida alcalina durante toda a benzoilação, e a temperatura foi mantida abaixo de 40 °C por arrefecimento. Quando o cloreto de benzoílo reagiu completamente, a mistura foi arrefecida e acidificada com 300 mL de ácido HCl concentrado. O produto em bruto foi filtrado e lavado com água, após o que foi extraído com 4 porções de 1 L de água quente para remover o ácido benzoico. O produto xaroposo quente (2), após decantação do extrato aquoso, foi cristalizado a partir de alguns volumes de metanol; rendimento 103 g (70 %), MP: 151-153 °C.
1-Benzoil-5-ceto-1,2,2a,3,4,5-hexa-hidrobenz-[cd]-indole (3)
1-Benzoil-3-(beta-carboxietil)-2,3-di-hidroindole (2), 118 g (0,4 mol), foi misturado com 200 mL de cloreto de tionilo puro. A solução foi deixada em repouso durante 30 minutos, após o que foi aquecida suavemente durante 15-20 minutos no banho de vapor. O excesso de cloreto de tionilo foi evaporado completamente abaixo de 30 °C no vácuo, e o cloreto de ácido em bruto foi dissolvido em 200 mL de dissulfureto de carbono. A solução de cloreto de ácido foi então adicionada numa corrente fina a uma suspensão vigorosamente agitada de 240 g de cloreto de alumínio em 1750 mL de dissulfureto de carbono contido num balão de 5 L (em HOOD!!!). Separou-se um complexo e a agitação tornou-se difícil. A mistura foi aquecida sob refluxo e agitada durante uma hora para completar a reação, após o que foi decomposta com muito cuidado pela adição de 500 g de gelo, 250 mL de ácido HCl conc. e 500 mL de água. Durante a decomposição, a agitação foi mantida e o arrefecimento foi afetado por destilação periódica do dissulfureto de carbono no vácuo, tendo o produto sido extraído com 2 L de benzeno. O extrato foi lavado cuidadosamente com 500 ml de hidróxido de sódio 2N em três porções e depois com água. Secou-se sobre sulfato de magnésio e evaporou-se a um pequeno volume no vácuo. A adição lenta de vários volumes de éter provocou a cristalização da cetona amarela (3) . Foi filtrada e lavada com éter; rendimento 85,3 g (77 %), MP: 146-147 °C. Uma amostra foi recristalizada para análise a partir de éter de benzeno.
1-Benzoil-3-(beta-carboxietil)-2,3-di-hidroindole (2), 118 g (0,4 mol), foi misturado com 200 mL de cloreto de tionilo puro. A solução foi deixada em repouso durante 30 minutos, após o que foi aquecida suavemente durante 15-20 minutos no banho de vapor. O excesso de cloreto de tionilo foi evaporado completamente abaixo de 30 °C no vácuo, e o cloreto de ácido em bruto foi dissolvido em 200 mL de dissulfureto de carbono. A solução de cloreto de ácido foi então adicionada numa corrente fina a uma suspensão vigorosamente agitada de 240 g de cloreto de alumínio em 1750 mL de dissulfureto de carbono contido num balão de 5 L (em HOOD!!!). Separou-se um complexo e a agitação tornou-se difícil. A mistura foi aquecida sob refluxo e agitada durante uma hora para completar a reação, após o que foi decomposta com muito cuidado pela adição de 500 g de gelo, 250 mL de ácido HCl conc. e 500 mL de água. Durante a decomposição, a agitação foi mantida e o arrefecimento foi afetado por destilação periódica do dissulfureto de carbono no vácuo, tendo o produto sido extraído com 2 L de benzeno. O extrato foi lavado cuidadosamente com 500 ml de hidróxido de sódio 2N em três porções e depois com água. Secou-se sobre sulfato de magnésio e evaporou-se a um pequeno volume no vácuo. A adição lenta de vários volumes de éter provocou a cristalização da cetona amarela (3) . Foi filtrada e lavada com éter; rendimento 85,3 g (77 %), MP: 146-147 °C. Uma amostra foi recristalizada para análise a partir de éter de benzeno.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
1-Benzoil-2,2a,3,4-tetrahidro-4-[metil-(2-metil-1,2-dioxolan-2-il-metil)-amino]-benz-[cd]-indol-5-(1H)-ona (6)
Uma solução de 270 g (0,76 mol) de 1-benzoil-4-bromo-5-ceto-1,2,2a,3,4,5-hexa-hidrobenz-[cd]-indole(4) e 307 g (2.35 mol) de metilaminoacetona etileno-cetal (5) em 4500 mL de benzeno seco foram refluxados sob azoto durante 21 h em 10 L RBF com condensador de refluxo. A mistura foi arrefecida e 151 g (93,5 %) de bromidrato de metilaminoacetona etileno-cetal foram filtrados, MP: 158-159 °C.
O filtrado foi lavado várias vezes com água gelada, após o que foi extraído com 2,5 L de ácido HCl diluído frio, contendo 150 mL do ácido concentrado. Os extractos ácidos foram imediatamente adicionados a um excesso de hidróxido de sódio diluído e gelado. O produto foi extraído com 1 L de clorofórmio e a solução de clorofórmio foi seca sobre sulfato de magnésio, tratada com carbono e concentrada no vácuo. A cetal-cetona residual (6) foi cristalizada a partir de acetona; MP: e mistura MP: 135-136 °C, com um rendimento de 220 g (71 %).
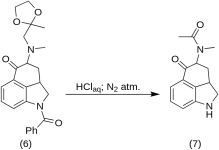
5-Keto-4-[N-metil-N-acetonilamino]-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (7)
20 g de 1-benzoil-2,2a,3,4-tetrahidro-4-[metil-(2-metil-1,3-dioxolan-2-il-metil)-amino]-benz-[cd]-indol-5-(1H)-ona (6) foram dissolvidos numa mistura de 250 mL de ácido HCl concentrado e 250 mL de água, tendo a solução sido mantida sob azoto a 37 °C em 3-5 L de RBF durante cinco dias. A mistura foi arrefecida, tratada com carvão, filtrada e o filtrado foi concentrado no vácuo até um pequeno volume. O resíduo foi tratado com excesso de bicarbonato de sódio; o produto foi extraído com clorofórmio frio e o solvente foi removido no vácuo à temperatura ambiente. A dicetona em bruto (7) foi pulverizada, misturada com cerca de 75 mL de éter benzénico 1:1 e filtrada; rendimento 9,8 g (77 %), MP: 105-107 °C. Uma amostra para análise foi recristalizada a partir de éter de benzeno ou etanol; MP: 109-110 °C; um monocloridrato foi obtido a partir de etanol diluído; MP: 200 °C dec.
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (8)
25 g de 5-Keto-4-[N-metil-N-acetonil]-amino-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (7) foram misturados com 550 mL de etanol absoluto. A mistura foi agitada sob azoto e arrefecida a -15 °C em 2-5 L de RBF. Adicionou-se então metóxido de sódio, 16,9 g, e a mistura foi agitada entre -10 °C e -12 °C durante 10 minutos. A mistura reacional foi arrefecida a -25 °C e o produto foi filtrado num funil de Buchner de 6,5 polegadas e lavado com um pouco de etanol frio e éter. Com uma exposição mínima ao ar (contém metóxido de sódio!), a cetona bruta (8) foi imediatamente agitada com um pouco de água gelada e filtrada de novo. Foi lavada com água gelada, etanol e éter; rendimento 16,2 g (69 %), MP: 145-147 °C. Uma amostra analítica foi recristalizada a partir de etanol diluído; MP: 155-157 °C; O dicloridrato foi preparado e recristalizado a partir de acetona aquosa; MP: 270 °C dec.
4-Acetil-9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (9)
9-Ceto-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (8), 24 g, foram adicionados a 80 mL de anidrido acético frio. A mistura foi mantida a 25 °C em 200 mL de RBF durante cerca de 5 min, após o que foi completamente arrefecida, e o produto (9) foi filtrado e lavado com éter; rendimento 20,5 g (76 %), mp: 167-170 °C. Obteve-se uma segunda colheita por evaporação do filtrado, o que aumentou o rendimento total para 82%. Uma amostra foi recristalizada a partir de acetona-etanol; MP: 169-170 °C; O cloridrato foi preparado em etanol e foi recristalizado a partir de etanol aquoso; MP: 250 °C dec.
9-Ceto-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (8), 24 g, foram adicionados a 80 mL de anidrido acético frio. A mistura foi mantida a 25 °C em 200 mL de RBF durante cerca de 5 min, após o que foi completamente arrefecida, e o produto (9) foi filtrado e lavado com éter; rendimento 20,5 g (76 %), mp: 167-170 °C. Obteve-se uma segunda colheita por evaporação do filtrado, o que aumentou o rendimento total para 82%. Uma amostra foi recristalizada a partir de acetona-etanol; MP: 169-170 °C; O cloridrato foi preparado em etanol e foi recristalizado a partir de etanol aquoso; MP: 250 °C dec.
4-Acetil-9-hidroxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (10)
10 g de 4-acetil-9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octahidroxindolo-[4,3-fg]-quinolina (9) foram adicionados a uma mistura de 150 mL de metanol e 10 mL de água em 500 mL de RBF. Adicionou-se borohidreto de sódio, 1,5 g, e deixou-se a reação prosseguir à temperatura ambiente até um pequeno volume, e adicionou-se uma mistura de 15 mL de ácido HCl conc. e 60 mL de água. O cloridrato (10) que se separou no arrefecimento foi filtrado e lavado com metanol, 9,0 g (79 %). Uma amostra foi recristalizada a partir de etanol diluído; MP: 245-246 °C dec.
Cloridrato de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (11)
Cloridrato de 4-acetil-9-hidroxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (10), 3.1 g, foi dissolvido em 75 mL de dióxido de enxofre líquido contido num invólucro de vidro numa autoclave de aço de 500 mL. Foi adicionado cloreto de tionilo, 1,2 mL, e o recipiente foi selado e mantido a 25 °C durante 6 h. O autoclave foi ventilado e a mistura de reação foi removida. O dióxido de enxofre foi deixado evaporar enquanto o volume da solução foi mantido constante pela adição lenta de éter seco. O cloridrato de cloro amorfo (11) foi filtrado, lavado com éter e seco no vácuo. MP: 130-135 °C dec. Rendimento: 3,5 g. A utilização do álcool 9-beta-epimérico nesta reação produziu o mesmo cloreto com um rendimento comparável.
4-Acetil-9-ciano-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (12)
Adicionaram-se 40 g de cianeto de sódio seco e em pó a 300 ml de cianeto de hidrogénio líquido gelado. A mistura foi agitada e arrefecida em gelo, tendo sido adicionados 7,5 g do cloridrato amorfo em bruto de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo [4,3f/g]-quinolina (11) acima referido. A agitação foi mantida em 500 ml de RBF durante 30 minutos, após o que o cianeto de hidrogénio foi rapidamente destilado sob pressão reduzida a cerca de 10 °C. O resíduo foi misturado com clorofórmio e água gelada, e a mistura resultante foi filtrada. A camada orgânica foi separada e a fase aquosa foi extraída duas vezes com clorofórmio. Os extractos combinados foram secos sobre sulfato de magnésio, descolorizados e o solvente foi destilado no vácuo. O produto (12) foi cristalizado a partir de acetato de etilo; rendimento de 3,3 g (54% do total com base no cloridrato de álcool), m.p. 172-174 °C. Arecristalização a partir do mesmo solvente aumentou o p.m. para 181-182 °C.
Adicionaram-se 40 g de cianeto de sódio seco e em pó a 300 ml de cianeto de hidrogénio líquido gelado. A mistura foi agitada e arrefecida em gelo, tendo sido adicionados 7,5 g do cloridrato amorfo em bruto de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo [4,3f/g]-quinolina (11) acima referido. A agitação foi mantida em 500 ml de RBF durante 30 minutos, após o que o cianeto de hidrogénio foi rapidamente destilado sob pressão reduzida a cerca de 10 °C. O resíduo foi misturado com clorofórmio e água gelada, e a mistura resultante foi filtrada. A camada orgânica foi separada e a fase aquosa foi extraída duas vezes com clorofórmio. Os extractos combinados foram secos sobre sulfato de magnésio, descolorizados e o solvente foi destilado no vácuo. O produto (12) foi cristalizado a partir de acetato de etilo; rendimento de 3,3 g (54% do total com base no cloridrato de álcool), m.p. 172-174 °C. Arecristalização a partir do mesmo solvente aumentou o p.m. para 181-182 °C.
9-Carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octahidroindolo-[4,3-fg]-quinolina (13)
O produto (12) acima, 1,0 g, foi misturado com 15 mL de metanol e 0,25 mL de água. A mistura foi arrefecida e adicionou-se lentamente 2 mL de ácido sulfúrico concentrado. A solução foi selada num tubo de vidro sob azoto e aquecida a 100 °C durante 23 a 24 h num RBF de 100 mL com condensador de refluxo. A mistura foi tratada com carbono descolorido e depois concentrada no vácuo para cerca de 10 mL. Foi vertida numa mistura de clorofórmio (30 mL), gelo e 10 g de bicarbonato de sódio. A camada de clorofórmio foi separada, e a fase aquosa foi extraída com 3 porções de 10 mL de clorofórmio. Os extractos combinados foram secos sobre sulfato de magnésio, evaporados até à secura, e o produto (13) foi cristalizado a partir de benzeno; rendimento 0,51 g (53 %), MP: 159-160 °C. Foi recristalizado a partir de acetato de etilo; MP: 160-161 °C.
O produto (12) acima, 1,0 g, foi misturado com 15 mL de metanol e 0,25 mL de água. A mistura foi arrefecida e adicionou-se lentamente 2 mL de ácido sulfúrico concentrado. A solução foi selada num tubo de vidro sob azoto e aquecida a 100 °C durante 23 a 24 h num RBF de 100 mL com condensador de refluxo. A mistura foi tratada com carbono descolorido e depois concentrada no vácuo para cerca de 10 mL. Foi vertida numa mistura de clorofórmio (30 mL), gelo e 10 g de bicarbonato de sódio. A camada de clorofórmio foi separada, e a fase aquosa foi extraída com 3 porções de 10 mL de clorofórmio. Os extractos combinados foram secos sobre sulfato de magnésio, evaporados até à secura, e o produto (13) foi cristalizado a partir de benzeno; rendimento 0,51 g (53 %), MP: 159-160 °C. Foi recristalizado a partir de acetato de etilo; MP: 160-161 °C.
Ácido dl-lisérgico sintético (14)
Uma mistura de 9-carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (13), 3,9 g, e 78 mL de solução de hidróxido de potássio a 1,5% foi refluxada durante 30 minutos sob azoto. Adicionou-se arseniato de sódio hidratado, 8,5 g, e níquel Raney (16 g, húmido), previamente desativado por ebulição em suspensão de xileno, e a mistura foi aquecida sob refluxo e agitada numa atmosfera de azoto durante 20 horas em 200 mL de RBF com condensador de refluxo. A solução foi tratada com carbono, e o ácido lisérgico bruto (14) foi precipitado por neutralização a pH 5,6. Foi filtrado e lavado com água; rendimento 1,04 g, MP: 240-242 °C dec. Foi também obtida uma segunda colheita, 0,16 g, MP: 233-235 °C dec.; rendimento total 30%. Oácido pode ser purificado dissolvendo-o em hidróxido de amónio diluído, tratando-o com carbono descolorante e reprecipitando-o com dióxido de carbono, MP: 242-243 °C dec; uma mistura com ácido dl-lisérgico obtida a partir de ácido d-lisérgico natural foi igualmente 242-243 °C dec. O ácido anidro foi obtido por secagem no vácuo durante várias horas a 150 °C.
Uma mistura de 9-carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (13), 3,9 g, e 78 mL de solução de hidróxido de potássio a 1,5% foi refluxada durante 30 minutos sob azoto. Adicionou-se arseniato de sódio hidratado, 8,5 g, e níquel Raney (16 g, húmido), previamente desativado por ebulição em suspensão de xileno, e a mistura foi aquecida sob refluxo e agitada numa atmosfera de azoto durante 20 horas em 200 mL de RBF com condensador de refluxo. A solução foi tratada com carbono, e o ácido lisérgico bruto (14) foi precipitado por neutralização a pH 5,6. Foi filtrado e lavado com água; rendimento 1,04 g, MP: 240-242 °C dec. Foi também obtida uma segunda colheita, 0,16 g, MP: 233-235 °C dec.; rendimento total 30%. Oácido pode ser purificado dissolvendo-o em hidróxido de amónio diluído, tratando-o com carbono descolorante e reprecipitando-o com dióxido de carbono, MP: 242-243 °C dec; uma mistura com ácido dl-lisérgico obtida a partir de ácido d-lisérgico natural foi igualmente 242-243 °C dec. O ácido anidro foi obtido por secagem no vácuo durante várias horas a 150 °C.
Attachments

Last edited:
