
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,792
- Solutions
- 3
- Reaction score
- 3,053
- Points
- 113
- Deals
- 1
Въведение
Лизергиновата киселина, основният фрагмент, получен от алкалоидите на ергота, е синтезирана в четиринадесет последователности, започващи с 3-бета-карбоксиетилиндол. Изходният материал е превърнат в междинен продукт 1-бензоил-5-кето-1,2,2а-3,4,5-хексахидробенз-[cd]-индол (3), съдържащ три от четирите пръстена, присъстващи в лизергиновата киселина. Този кетон от своя страна се превръща в тетрацикличното съединение 9-кето-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4.3-fe]-хинолин (8), а оттам и в лизергинова киселина (14). Този синтез не е прост и изисква голям лабораторен опит и познания по химия. Освен това има няколко манипулации с опасни вещества, които трябва да се извършват при строги мерки за безопасност.
Температура на кипене: 536,2 ± 50,0 °C при 760 mm Hg;
Температура на топене: 240 °C;
Молекулно тегло: 268,31 g/mole;
Плътност: 1,4 ± 0,1 g/mL;
CAS номер: 82-58-6.
Оборудване и стъклария:
- Стоманен реактор за хидрогениране 2-3 л;
- Стоманен автоклав 500 mL;
- Лабораторна везна (0,01 - 500 g е подходяща);
- Кръглодънни колби 100, 200, 500 ml, 5 и 10 l;
- Компресор за водород (H2) и произход;
- Колба и фуния на Бюхнер (голяма) 5 L [филтърът на Шот може да се използва за малки количества];
- Ротационна машина (голяма);
- Източник на вакуум;
- Разделителни фунии от 500 ml и 2 L;
- Азотен балон ~50-70 L (1 бар);
- Капачки за колби;
- Водна баня с осолена ледена вода;
- 5 L x2, 2 L x2; 1 L x2; 500 mL x2; 100 mL x3; 50 mL x2 Чаши;
- Стъклена спринцовка или пипета на Пастьор;
- Магнитна бъркалка или горна бъркалка;
- Инсталация за вакуумна дестилация;
- Възвратен кондензатор;
- Стойка за реторта и скоба за закрепване на апаратурата;
- Лабораторен термометър (от -20 °C до 200 °C) с адаптер за колба;
- Индикаторна хартия за рН;
- Стъклена пръчка и шпатула;
- 250-ватова крушка.
Реактиви.
- 3-индолепропионова киселина (1), 94,6 g (0,5 mol);
- 9,5 л дестилирана вода (H2O);
- ~400 g натриев хидроксид (NaOH);
- 116 g никел на Рани (Ni);
- 1050 mL концентрирана солна киселина (HCl);
- 2 mL сярна киселина (H2SO4);
- 210 mL 12N разтвор на натриев хидроксид (NaOH) aq;
- 180 mL бензоилхлорид;
- ~1,5 л метанол (MeOH);
- ~1,6 л етанол (EtOH);
- 201,2 mL тионилхлорид (SOCl2);
- 1950 mL въглероден дисулфид (CS2);
- 240 g Алуминиев хлорид (AlCl3);
- 2,5 L бензол;
- 500 mL 2N натриев хидроксид (NaOH);
- ~3,2 L диетил етер (Et2O);
- 3,3 L Ледена оцетна киселина (AcOH);
- 352 g (1,1 mol) Пиридин хидробромид пербромид;
- 5 L хлороформ (CHCl3);
- ~1000 g Магнезиев сулфат (MgSO4);
- 307 g (2,35 mol) Метиламиноацетон етиленкетал ( 5);
- 4,5 L бензол;
- ~500 g Активен въглен (С);
- ~1 L ацетон;
- ~500 g Натриев бикарбонат (NaHCO3);
- 80 mL Студен оцетен анхидрид (Ac2O);
- 1,5 g натриев борхидрид (NaBH4);
- 75 mL Серен диоксид (SO2 течност);
- 40 g натриев цианид (NaCN на прах);
- 300 mL Водороден цианид (HCN течност);
- 78 mL 1,5% разтвор на калиев хидроксид (KOH);
- 8,5 g хидратиран натриев арсенат;
- ~ 50 mL ксилол;
- 100 mL разреден разтвор на амониев хидроксид (NH4OH);
- 16,9 g натриев метоксид (MeONa).
Процедура
1-бензоил-3-(бета-карбоксиетил)-2,3-дихидроиндол (2)3-индолепропионова киселина (1), 94,6 g (0,5 mol), се разтваря в 600 mL вода, съдържаща 20 g натриев хидроксид. Разтворът се смесва с около 100 g никелов катализатор на Раней и се хидрогенира при RT в 2-3 L стоманена хидрогенираща бомба при налягане 3000-4000 psi (207-276 bar) H2. Редукцията обикновено завършва за 20-30 часа, след което катализаторът се филтрира и се промива с малко вода. Към филтрата се прибавя концентрирана HCl киселина, 85 ml, и разтворът се охлажда. Ако редукцията е била непълна, нереагиралата индолпропионова киселина се отделя в този момент и се отстранява чрез филтриране. След това филтратът се бензоилира по обичайната процедура на Шоттен-Бауман, като се използват 210 ml 12N натриев хидроксид и 180 ml бензоилхлорид. Разтворът е поддържан алкален по време на бензоелирането, а температурата е поддържана под 40 °C чрез охлаждане. Когато бензоилхлоридът е реагирал напълно, сместа е охладена и подкиселена с 300 mL концентрирана HCl киселина. Необработеният продукт се филтрира и се промива с вода, след което се екстрахира с 4 x 1 L порции гореща вода, за да се отстрани бензоената киселина. Горещият сиропиран продукт ( 2), след декантиране на водния екстракт, е кристализиран от няколко обема метанол; добив 103 g (70 %), MP: 151-153 °C.
1-бензоил-5-кето-1,2,2а,3,4,5-хексахидробенз-[cd]-индол (3)
1-бензоил-3-(бета-карбоксиетил)-2,3-дихидроиндол (2), 118 g (0,4 mol), се смесва с 200 ml чист тионилхлорид. Разтворът е оставен да престои 30 min, след което е загряван внимателно в продължение на 15-20 min на парна баня. Излишният тионилхлорид се изпарява напълно под 30 °C във вакуум, а суровият киселинен хлорид се разтваря в 200 ml въглероден дисулфид. След това разтворът на киселинния хлорид се прибавя на тънка струя към енергично разбърквана суспензия от 240 g алуминиев хлорид в 1750 mL въглероден дисулфид, намираща се в 5-литрова колба (в КАПАК!!!). Отделя се комплекс и разбъркването става трудно. Сместа се нагрява под обратен поток и се разбърква в продължение на един час, за да се завърши реакцията, след което се разгражда много внимателно, като се добавят 500 g лед, 250 mL конц. киселина HCl и 500 mL вода. По време на разлагането се поддържаше разбъркване, а охлаждането се повлияваше от периодичната дестилация на въглеродния дисулфид във вакуума и продуктът се екстрахираше с 2 L бензол. Екстрактът се промива старателно с 500 mL 2N натриев хидроксид на три порции и след това с вода. Изсушава се над магнезиев сулфат и след това се изпарява до малък обем във вакуума. Бавното добавяне на няколко обема етер води до кристализация на жълтия кетон (3 ). Той се филтрира и се промива с етер; добив 85,3 g (77 %), MP: 146-147 °C. Една проба е рекристализирана за анализ от бензен-етер.
1-бензоил-3-(бета-карбоксиетил)-2,3-дихидроиндол (2), 118 g (0,4 mol), се смесва с 200 ml чист тионилхлорид. Разтворът е оставен да престои 30 min, след което е загряван внимателно в продължение на 15-20 min на парна баня. Излишният тионилхлорид се изпарява напълно под 30 °C във вакуум, а суровият киселинен хлорид се разтваря в 200 ml въглероден дисулфид. След това разтворът на киселинния хлорид се прибавя на тънка струя към енергично разбърквана суспензия от 240 g алуминиев хлорид в 1750 mL въглероден дисулфид, намираща се в 5-литрова колба (в КАПАК!!!). Отделя се комплекс и разбъркването става трудно. Сместа се нагрява под обратен поток и се разбърква в продължение на един час, за да се завърши реакцията, след което се разгражда много внимателно, като се добавят 500 g лед, 250 mL конц. киселина HCl и 500 mL вода. По време на разлагането се поддържаше разбъркване, а охлаждането се повлияваше от периодичната дестилация на въглеродния дисулфид във вакуума и продуктът се екстрахираше с 2 L бензол. Екстрактът се промива старателно с 500 mL 2N натриев хидроксид на три порции и след това с вода. Изсушава се над магнезиев сулфат и след това се изпарява до малък обем във вакуума. Бавното добавяне на няколко обема етер води до кристализация на жълтия кетон (3 ). Той се филтрира и се промива с етер; добив 85,3 g (77 %), MP: 146-147 °C. Една проба е рекристализирана за анализ от бензен-етер.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
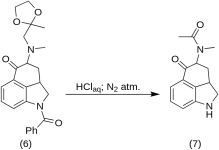
1-бензоил-2,2а,3,4-тетрахидро-4-[метил-(2-метил-1,2-диоксолан-2-ил-метил)-амино]-бенз-[cd]-индол-5-(1H)-он (6)
Разтвор на 270 g (0,76 mol) 1-бензоил-4-бромо-5-кето-1,2,2а,3,4,5-хексахидробенз-[cd]-индол(4) и 307 g (2.35 mol) на метиламиноацетон етилен кетал (5) в 4500 ml сух бензол се рефлуксира под азот в продължение на 21 h в 10 L RBF с обратен хладник. Сместа се охлажда и се филтрира 151 g (93,5 %) метиламиноацетон етиленкетал хидробромид, MP: 158-159 °C.
Филтратът се промива няколко пъти с ледена вода, след което се екстрахира с 2,5 L студена, разредена HCl киселина, съдържаща 150 ml от концентрираната киселина. Киселинните екстракти веднага се добавят към излишък от ледено студен разреден натриев хидроксид. Продуктът се екстрахира с 1 L хлороформ, а хлороформният разтвор се изсушава над магнезиев сулфат, третира се с въглерод и се концентрира във вакуум. Остатъчният кетал-кетон (6 ) е кристализиран от ацетон; MP: и смес MP: 135-136 °C, добив 220 g (71 %).
5-кето-4-[N-метил-N-ацетониламино]-1,2,2а,3,4,5-хексахидробенз-[кд]-индол (7)
20 g 1-бензоил-2,2а,3,4-тетрахидро-4-[метил-(2-метил-1,3-диоксолан-2-ил-метил)-амино]-бенз-[cd]-индол-5-(1H)-он (6) се разтваря в смес от 250 ml концентрирана HCl киселина и 250 ml вода и разтворът се съхранява под азот при 37 °C в 3-5 L RBF в продължение на пет дни. Сместа се охлажда, обработва се с въглерод, филтрира се и филтратът се концентрира във вакуум до малък обем. Остатъкът се третира с излишък от натриев бикарбонат; продуктът се екстрахира със студен хлороформ и разтворителят се отстранява във вакуума при RT. Необработеният дикетон (7 ) се стрива на прах, размива се с около 75 ml бензен-етер в съотношение 1:1 и се филтрира; добив 9,8 g (77 %), MP: 105-107 °C. Пробата за анализ е рекристализирана от бензен-етер или етанол; МР: 109-110 °C; Монохидрохлорид е получен от разреден етанол; МР: 200 °C дек.
9-Кето-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (8)
25 g 5-Кето-4-[N-метил-N-ацетонил]-амино-1,2,2а,3,4,5-хексахидробенз-[cd]-индол (7 ) се смесва с 550 mL абсолютен етанол. Сместа се разбърква под азот и се охлажда до -15 °C в 2-5 L RBF. След това се добавя натриев метоксид, 16,9 g, и сместа се разбърква при -10 °C до -12 °C в продължение на десет минути. Реакционната смес се охлажда до -25 °C, а продуктът се филтрира на 6,5-инчова бухнерска фуния и се промива с малко студен етанол и етер. При минимално излагане на въздух (съдържа натриев метоксид!) суровият кетон (8 ) веднага се размива с малко ледена вода и се филтрира отново. Той се промива с ледена вода, етанол и етер; добив 16,2 g (69 %), MP: 145-147 °C. Аналитична проба е рекристализирана от разреден етанол; МР: 155-157 °С; Дихидрохлоридът е приготвен и рекристализиран от воден ацетон; МР: 270 °С дек.
4-ацетил-9-кето-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (9)
9-кето-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (8), 24 g, се добавя към 80 mL студен оцетен анхидрид. Сместа се поддържа при 25 °С в 200 ml RBF за около 5 min, след което се охлажда добре и продуктът (9 ) се филтрира и промива с етер; добив 20,5 g (76 %), mp: 167-170 °C. Чрез изпаряване на филтрата е получена втора реколта, която повишава общия добив до 82 %. Една проба е рекристализирана от ацетон-етанол; МР: 169-170 °С; Хидрохлоридът е приготвен в етанол и е рекристализиран от воден етанол; МР: 250 °С дек.
9-кето-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (8), 24 g, се добавя към 80 mL студен оцетен анхидрид. Сместа се поддържа при 25 °С в 200 ml RBF за около 5 min, след което се охлажда добре и продуктът (9 ) се филтрира и промива с етер; добив 20,5 g (76 %), mp: 167-170 °C. Чрез изпаряване на филтрата е получена втора реколта, която повишава общия добив до 82 %. Една проба е рекристализирана от ацетон-етанол; МР: 169-170 °С; Хидрохлоридът е приготвен в етанол и е рекристализиран от воден етанол; МР: 250 °С дек.
4-ацетил-9-хидрокси-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (10)
10 g 4-ацетил-9-кето-7-метил-4,5,5а,6,6а,7,8,9-октахидроксиндоло-[4,3-fg]-хинолин (9 ) се добавя към смес от 150 ml метанол и 10 ml вода в 500 ml RBF. Добавя се натриев борохидрид, 1,5 g, и реакцията се оставя да протече при RT до малък обем, след което се добавя смес от 15 mL конц. киселина HCl и 60 mL вода. Хидрохлоридът ( 10 ), който се отделя при охлаждането, се филтрира и се промива с метанол, 9,0 g (79 %). Една проба е рекристализирала от разреден етанол; MP: 245-246 °C dec.
4-ацетил-9-хлоро-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин хидрохлорид (11)
4-ацетил-9-хидрокси-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин хидрохлорид (10), 3.1 g, се разтваря в 75 ml течен серен диоксид, поставен в стъклена обвивка в 500 ml стоманен автоклав. Добавя се 1,2 ml тионилхлорид, съдът се затваря и се държи при 25 °C в продължение на 6 h. Автоклавът се изпуска и реакционната смес се отстранява. Серният диоксид се оставя да се изпари, докато обемът на разтвора се поддържа постоянен чрез бавно добавяне на сух етер. Аморфният хлорхидрохлорид ( 11 ) се филтрира, промива се с етер и се изсушава във вакуум, MP: 130-135 °C dec. Добив 3,5 g. Използването на 9-бета-епимерен алкохол в тази реакция дава същия хлорид със сравним добив.
4-ацетил-9-циано-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (12)
Сух, прахообразен натриев цианид, 40 g, се добавя към 300 ml ледено студен течен циановодород. Сместа се разбърква и охлажда в лед, след което се добавят 7,5 g от суровия аморфен 4-ацетил-9-хлор-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло[4,3f/g]-хинолин хидрохлорид ( 11) по-горе. Разбъркването продължава в 500 ml RBF за 30 min, след което циановодородът бързо се дестилира при понижено налягане под около 10 °C. Остатъкът се смесва с хлороформ и ледена вода и получената смес се филтрира. Органичният слой е отделен, а водната фаза е екстрахирана два пъти с хлороформ. Комбинираните екстракти се изсушават над магнезиев сулфат, обезцветяват се и разтворителят се дестилира във вакуум. Продуктът (12 ) е кристализиран от етилов ацетат; добив 3,3 g (54 % от общото количество на базата на алкохолния хидрохлорид), m.p. 172-174 °C. Повторната кристализация от същия разтворител повишава m.p. до 181-182 °C.
Сух, прахообразен натриев цианид, 40 g, се добавя към 300 ml ледено студен течен циановодород. Сместа се разбърква и охлажда в лед, след което се добавят 7,5 g от суровия аморфен 4-ацетил-9-хлор-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло[4,3f/g]-хинолин хидрохлорид ( 11) по-горе. Разбъркването продължава в 500 ml RBF за 30 min, след което циановодородът бързо се дестилира при понижено налягане под около 10 °C. Остатъкът се смесва с хлороформ и ледена вода и получената смес се филтрира. Органичният слой е отделен, а водната фаза е екстрахирана два пъти с хлороформ. Комбинираните екстракти се изсушават над магнезиев сулфат, обезцветяват се и разтворителят се дестилира във вакуум. Продуктът (12 ) е кристализиран от етилов ацетат; добив 3,3 g (54 % от общото количество на базата на алкохолния хидрохлорид), m.p. 172-174 °C. Повторната кристализация от същия разтворител повишава m.p. до 181-182 °C.
9-Карбометокси-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (13)
Точно посоченият по-горе продукт (12) , 1,0 g, се смесва с 15 ml метанол и 0,25 ml вода. Сместа се охлажда и бавно се добавят 2 mL концентрирана сярна киселина. Разтворът е запечатан в стъклена епруветка под азот и се нагрява при 100 °С в продължение на 23 до 24 h в 100 mL RBF с обратен хладник. Сместа се обработва с обезцветен въглерод и след това се концентрира във вакуума до около 10 ml. Тя се излива върху смес от хлороформ (30 mL), лед и 10 g натриев бикарбонат. Слоят хлороформ е отделен, а водната фаза е екстрахирана с 3 x 10 ml порции хлороформ. Комбинираните екстракти се изсушават над магнезиев сулфат, изпаряват се до сухост и продуктът (13 ) кристализира от бензол; добив 0,51 g (53 %), MP: 159-160 °C. Той е повторно кристализиран от етилов ацетат; MP: 160-161 °C.
Точно посоченият по-горе продукт (12) , 1,0 g, се смесва с 15 ml метанол и 0,25 ml вода. Сместа се охлажда и бавно се добавят 2 mL концентрирана сярна киселина. Разтворът е запечатан в стъклена епруветка под азот и се нагрява при 100 °С в продължение на 23 до 24 h в 100 mL RBF с обратен хладник. Сместа се обработва с обезцветен въглерод и след това се концентрира във вакуума до около 10 ml. Тя се излива върху смес от хлороформ (30 mL), лед и 10 g натриев бикарбонат. Слоят хлороформ е отделен, а водната фаза е екстрахирана с 3 x 10 ml порции хлороформ. Комбинираните екстракти се изсушават над магнезиев сулфат, изпаряват се до сухост и продуктът (13 ) кристализира от бензол; добив 0,51 g (53 %), MP: 159-160 °C. Той е повторно кристализиран от етилов ацетат; MP: 160-161 °C.
Синтетична dl-лизергинова киселина (14)
Смес от 9-карбометокси-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (13), 3,9 g, и 78 ml 1,5 % разтвор на калиев хидроксид се рефлуксира в продължение на 30 min под азот. Добавят се хидратиран натриев арсенат, 8,5 g, и никел на Раней (16 g, мокър), предварително деактивиран чрез варене в суспензия от ксилол, и сместа се нагрява под обратен хладник и се разбърква в азотна атмосфера в продължение на 20 часа в 200 mL RBF с обратен хладник. Разтворът е третиран с въглерод и суровата лизергинова киселина (14 ) е утаена чрез неутрализация до pH 5,6. Тя е филтрирана и промита с вода; добив 1,04 g, MP: 240-242 °C dec. Получена е и втора култура, 0,16 g, MP: 233-235 °C dec.; общ добив 30 %. Киселината може да бъде пречистена чрез разтваряне в разреден амониев хидроксид, третиране с обезцветяващ въглен и повторно утаяване с въглероден диоксид, MP: 242-243 °C dec; смес с dl-лизергинова киселина, направена от естествена d-лизергинова киселина, също е 242-243 °C dec. Безводната киселина се получава чрез сушене във вакуума за няколко часа при 150 °C.
Смес от 9-карбометокси-7-метил-4,5,5а,6,6а,7,8,9-октахидроиндоло-[4,3-fg]-хинолин (13), 3,9 g, и 78 ml 1,5 % разтвор на калиев хидроксид се рефлуксира в продължение на 30 min под азот. Добавят се хидратиран натриев арсенат, 8,5 g, и никел на Раней (16 g, мокър), предварително деактивиран чрез варене в суспензия от ксилол, и сместа се нагрява под обратен хладник и се разбърква в азотна атмосфера в продължение на 20 часа в 200 mL RBF с обратен хладник. Разтворът е третиран с въглерод и суровата лизергинова киселина (14 ) е утаена чрез неутрализация до pH 5,6. Тя е филтрирана и промита с вода; добив 1,04 g, MP: 240-242 °C dec. Получена е и втора култура, 0,16 g, MP: 233-235 °C dec.; общ добив 30 %. Киселината може да бъде пречистена чрез разтваряне в разреден амониев хидроксид, третиране с обезцветяващ въглен и повторно утаяване с въглероден диоксид, MP: 242-243 °C dec; смес с dl-лизергинова киселина, направена от естествена d-лизергинова киселина, също е 242-243 °C dec. Безводната киселина се получава чрез сушене във вакуума за няколко часа при 150 °C.
Attachments

Last edited:
